Dostinex 0.25mg, 0.5mg Cabergoline Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Dostinex 0,25 mg y cómo se usa?
Dostinex (cabergolina) es un antagonista de los receptores de dopamina que se usa para tratar un desequilibrio hormonal en el que hay demasiada prolactina en la sangre (también llamada hiperprolactinemia).
¿Cuáles son los efectos secundarios de Dostinex?
Los efectos secundarios comunes de Dostinex 0.25mg incluyen:
- náuseas,
- vómitos,
- malestar estomacal o dolor,
- indigestión,
- estreñimiento,
- gas,
- mareo,
- sensación de giro,
- aturdimiento,
- somnolencia,
- nerviosismo,
- cansancio,
- dolor de cabeza,
- estado de ánimo deprimido,
- Sofocos,
- entumecimiento o sensación de hormigueo, o
- boca seca.
Informe a su médico si experimenta efectos secundarios raros pero graves de Dostinex, incluidos:
- dificultad para respirar,
- tos persistente,
- hinchazón de tobillos o pies,
- cansancio inusual,
- cambios mentales/anímicos (por ejemplo, nerviosismo),
- impulsos fuertes e inusuales (por ejemplo, aumento de los juegos de azar, aumento de los impulsos sexuales),
- cambios de visión,
- menstruaciones dolorosas, o
- dolor en los senos.
DESCRIPCIÓN
Los comprimidos de DOSTINEX contienen cabergolina, un agonista del receptor de dopamina. El nombre químico de la cabergolina es 1-[(6-alilergolina-8β-il)-carbonil]-1-[3-(dimetilamino)propil]-3-etilurea. Su fórmula empírica es C26H37N5O2 y su peso molecular es 451,62. La fórmula estructural es la siguiente:
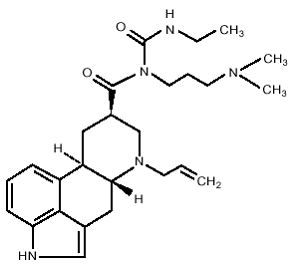
La cabergolina es un polvo blanco soluble en alcohol etílico, cloroformo y N, N-dimetilformamida (DMF); ligeramente soluble en ácido clorhídrico 0,1 N; muy poco soluble en n-hexano; e insoluble en agua.
Los comprimidos de DOSTINEX 0,25 mg, para administración oral, contienen 0,5 mg de cabergolina. Los ingredientes inactivos consisten en leucina, USP y lactosa, NF.
INDICACIONES
DOSTINEX 0,5 mg Comprimidos está indicado para el tratamiento de trastornos hiperprolactinémicos, ya sea idiopáticos o debidos a adenomas hipofisarios.
DOSIFICACIÓN Y ADMINISTRACIÓN
La dosis recomendada de DOSTINEX 0,25 mg comprimidos para el inicio del tratamiento es de 0,25 mg dos veces por semana. La dosis puede aumentarse en 0,25 mg dos veces por semana hasta una dosis de 1 mg dos veces por semana según el nivel de prolactina sérica del paciente. Antes de iniciar el tratamiento, se debe realizar una evaluación cardiovascular y se debe considerar una ecocardiografía para evaluar la enfermedad valvular.
Los aumentos de dosis no deben ocurrir más rápido que cada 4 semanas, para que el médico pueda evaluar la respuesta del paciente a cada nivel de dosis. Si el paciente no responde adecuadamente y no se observa ningún beneficio adicional con dosis más altas, se debe usar la dosis más baja que logró la respuesta máxima y se deben considerar otros enfoques terapéuticos. Los pacientes que reciben tratamiento a largo plazo con DOSTINEX 0,25 mg deben someterse a una evaluación periódica de su estado cardíaco y se debe considerar una ecocardiografía.
Después de que se haya mantenido un nivel de prolactina sérica normal durante 6 meses, se puede interrumpir el tratamiento con DOSTINEX 0,5 mg, con un control periódico del nivel de prolactina sérica para determinar si se debe reiniciar el tratamiento con DOSTINEX o cuándo. No se ha establecido la durabilidad de la eficacia más allá de los 24 meses de tratamiento con DOSTINEX.
CÓMO SUMINISTRADO
Los comprimidos de DOSTINEX son comprimidos blancos ranurados con forma de cápsula que contienen 0,5 mg de cabergolina. Cada tableta está ranurada en un lado y tiene la letra P y la letra U a ambos lados de la línea de corte. El otro lado de la tableta está grabado con el número 700.
DOSTINEX está disponible de la siguiente manera:
Frascos de 8 tabletas CDN 0013-7001-12
Almacenamiento
Almacenar a temperatura ambiente controlada de 20° a 25°C (68° a 77°F) [ver USP ].
La etiqueta de este producto puede haber sido actualizada. Para obtener información de prescripción completa y actualizada, visite www.pfizer.com
Distribuido por: Pharmacia & Upjohn Company Division of Pfizer Inc., NY, NY 10017. Revisado: septiembre de 2014
EFECTOS SECUNDARIOS
La seguridad de DOSTINEX 0.25mg Comprimidos ha sido evaluada en más de 900 pacientes con trastornos hiperprolactinémicos. La mayoría de los eventos adversos fueron de gravedad leve o moderada.
En un estudio doble ciego controlado con placebo de 4 semanas, el tratamiento consistió en placebo o cabergolina en dosis fijas de 0,125, 0,5, 0,75 o 1,0 mg dos veces por semana. Las dosis se redujeron a la mitad durante la primera semana. Dado que se observó un posible efecto relacionado con la dosis solo para las náuseas, se combinaron los cuatro grupos de tratamiento con cabergolina. La incidencia de los eventos adversos más comunes durante el estudio controlado con placebo se presenta en la siguiente tabla.
Incidencia de eventos adversos informados durante el ensayo doble ciego controlado con placebo de 4 semanas
En el período doble ciego de 8 semanas del ensayo comparativo con bromocriptina, DOSTINEX (a una dosis de 0,5 mg dos veces por semana) se suspendió debido a un evento adverso en 4 de 221 pacientes (2 %), mientras que la bromocriptina (a una dosis de 2,5 mg dos veces al día) se suspendió en 14 de 231 pacientes (6%). Las razones más comunes para la discontinuación de DOSTINEX fueron dolor de cabeza, náuseas y vómitos (3, 2 y 2 pacientes respectivamente); las razones más comunes para la interrupción de la bromocriptina fueron náuseas, vómitos, dolor de cabeza y mareos o vértigo (10, 3, 3 y 3 pacientes respectivamente). La incidencia de los eventos adversos más comunes durante la parte doble ciego del ensayo comparativo con bromocriptina se presenta en la siguiente tabla.
Incidencia de eventos adversos informados durante el período doble ciego de 8 semanas del ensayo comparativo con bromocriptina
A continuación se presentan otros eventos adversos que se informaron con una incidencia de
Cuerpo como un todo: edema facial, síntomas gripales, malestar general
Sistema cardiovascular: hipotensión, síncope, palpitaciones
Sistema digestivo: boca seca, flatulencia, diarrea, anorexia
Sistema metabólico y nutricional: pérdida de peso, aumento de peso
Sistema nervioso: somnolencia, nerviosismo, parestesia, insomnio, ansiedad
Sistema respiratorio: congestión nasal, epistaxis
Piel y apéndices: acné, prurito
Sentidos especiales: visión anormal
Sistema urogenital: dismenorrea, aumento de la libido
La seguridad de la cabergolina se ha evaluado en aproximadamente 1.200 pacientes con enfermedad de Parkinson en estudios controlados y no controlados a dosis de hasta 11,5 mg/día, lo que supera con creces la dosis máxima recomendada de cabergolina para los trastornos hiperprolactinémicos. Además de los eventos adversos que ocurrieron en pacientes con trastornos hiperprolactinémicos, los eventos adversos más comunes en pacientes con enfermedad de Parkinson fueron discinesia, alucinaciones, confusión y edema periférico. En raras ocasiones se produjeron insuficiencia cardíaca, derrame pleural, fibrosis pulmonar y úlcera gástrica o duodenal. Se ha notificado un caso de pericarditis constrictiva.
Datos de vigilancia posteriores a la comercialización
Se han informado los siguientes eventos en asociación con DOSTINEX: valvulopatía cardíaca y reacciones fibróticas extracardíacas, (Ver ADVERTENCIAS , Valvulopatía cardíaca y reacciones fibróticas extracardíacas ).
Se han informado otros eventos en asociación con cabergolina: hipersexualidad, aumento de la libido y juego patológico (Ver PRECAUCIONES , Psiquiátrico ). Además, se han informado casos de alopecia, agresión y trastorno psicótico en pacientes que toman DOSTINEX. Algunos de estos informes han sido en pacientes que han tenido reacciones adversas previas a productos agonistas de dopamina.
INTERACCIONES CON LA DROGAS
DOSTINEX 0,5 mg no debe administrarse simultáneamente con antagonistas D, como fenotiazinas, butirofenonas, tioxantenos o metoclopramida.
ADVERTENCIAS
El embarazo
Los agonistas de la dopamina en general no deben usarse en pacientes con hipertensión inducida por el embarazo, por ejemplo, preeclampsia, eclampsia e hipertensión posparto, a menos que se considere que el beneficio potencial supera el posible riesgo.
Complicaciones fibróticas
Valvulopatía cardíaca
Todos los pacientes deben someterse a una evaluación cardiovascular, incluido un ecocardiograma para evaluar la posible presencia de enfermedad valvular. Si se detecta enfermedad valvular, el paciente no debe ser tratado con DOSTINEX. (Ver CONTRAINDICACIONES ) Se han informado casos posteriores a la comercialización de valvulopatía cardíaca en pacientes que recibieron DOSTINEX. Estos casos generalmente han ocurrido durante la administración de dosis altas de DOSTINEX (> 2 mg/día) para el tratamiento de la enfermedad de Parkinson. También se informaron casos de valvulopatía cardíaca en pacientes que recibieron dosis más bajas de DOSTINEX para el tratamiento de trastornos hiperprolactinémicos.
Se llevó a cabo un estudio de cohorte retrospectivo de varios países que utilizó registros de práctica general y sistemas de enlace de registros en el Reino Unido, Italia y los Países Bajos para evaluar la asociación entre el nuevo uso de agonistas de la dopamina, incluida la cabergolina (n = 27 812) para la enfermedad de Parkinson y la hiperprolactinemia y los trastornos cardíacos. regurgitación valvular (RCV), otras fibrosis y otros eventos cardiopulmonares durante un máximo de 12 años de seguimiento. En este estudio, el uso de cabergolina entre personas con enfermedad de Parkinson se asoció con un mayor riesgo de RCV en comparación con los agonistas dopaminérgicos (AD) no derivados de la ergotamina y la levodopa [Tasa de incidencia (IR) por 10.000 años-persona de 68,1 (95 % de intervalo de confianza (IC): 37,2–115,3) para cabergolina frente a 10,0 (IC 95 %: 5,2–19,4) para AD sin cornezuelo y 11,3 (IC 95 %: 7,2–17,0) para levodopa]. En el análisis del estudio limitado a las personas con hiperprolactinemia tratada con agonistas de la dopamina (n=8386), en comparación con las que no la usaban (n=15,147), las personas expuestas a la cabergolina no tenían un riesgo elevado de RCV. Los resultados con respecto al riesgo de RCV asociado con el tratamiento con cabergolina para personas con enfermedad de Parkinson (riesgo aumentado) y aquellos con hiperprolactinemia (sin riesgo aumentado) son consistentes con los hallazgos de otros estudios publicados.
Los médicos deben usar la dosis efectiva más baja de DOSTINEX para el tratamiento de trastornos hiperprolactinémicos y deben reevaluar periódicamente la necesidad de continuar la terapia con DOSTINEX. Después del inicio del tratamiento, se debe realizar un seguimiento clínico y de diagnóstico (por ejemplo, radiografía de tórax, tomografía computarizada y ecocardiograma cardíaco) para evaluar el riesgo de valvulopatía cardíaca. La frecuencia recomendada de monitoreo ecocardiográfico de rutina es cada 6 a 12 meses o según esté clínicamente indicado ante la presencia de signos y síntomas como edema, nuevo soplo cardíaco, disnea o insuficiencia cardíaca congestiva.
DOSTINEX debe suspenderse si un ecocardiograma revela una nueva regurgitación valvular, restricción valvular o engrosamiento de las valvas.
DOSTINEX debe usarse con precaución en pacientes expuestos a otros medicamentos asociados con valvulopatía.

Reacciones fibróticas extracardíacas
Se han informado casos posteriores a la comercialización de fibrosis pleural, pericárdica y retroperitoneal después de la administración de DOSTINEX. Algunos informes se realizaron en pacientes tratados previamente con otros agonistas dopaminérgicos ergotínicos. DOSTINEX no debe utilizarse en pacientes con antecedentes de trastornos fibróticos cardíacos o extracardíacos.
Los trastornos fibróticos pueden tener un inicio insidioso y los pacientes deben ser monitoreados para detectar manifestaciones de fibrosis progresiva. Por lo tanto, durante el tratamiento, se debe prestar atención a los signos y síntomas de:
- Enfermedad pleuropulmonar como disnea, dificultad para respirar, tos persistente o dolor torácico.
- Insuficiencia renal u obstrucción vascular ureteral/abdominal que puede ocurrir con dolor en el lomo/flanco y edema en las extremidades inferiores, así como cualquier posible masa abdominal o dolor a la palpación que puede indicar fibrosis retroperitoneal.
- Insuficiencia cardíaca: los casos de fibrosis valvular y pericárdica a menudo se han manifestado como insuficiencia cardíaca. Por lo tanto, la fibrosis valvular (y la pericarditis constrictiva) deben descartarse si se presentan tales síntomas.
El monitoreo clínico y de diagnóstico, como la velocidad de sedimentación de eritrocitos, la radiografía de tórax, las mediciones de creatinina sérica y otras investigaciones deben considerarse al inicio del estudio y según sea necesario mientras los pacientes reciben tratamiento con DOSTINEX.
Luego del diagnóstico de derrame pleural o fibrosis pulmonar, se informó que la interrupción de DOSTINEX resultó en una mejoría de los signos y síntomas.
PRECAUCIONES
General
Las dosis iniciales superiores a 1,0 mg pueden producir hipotensión ortostática. Se debe tener cuidado al administrar DOSTINEX con otros medicamentos que se sabe que reducen la presión arterial.
Inhibición o supresión de la lactancia posparto
DOSTINEX 0,25 mg no está indicado para la inhibición o supresión de la lactancia fisiológica. El uso de bromocriptina, otro agonista de la dopamina para este propósito, se ha asociado con casos de hipertensión, accidente cerebrovascular y convulsiones.
Deterioro hepático
Dado que la cabergolina se metaboliza extensamente en el hígado, se debe tener precaución y se debe realizar un seguimiento cuidadoso cuando se administre DOSTINEX 0,5 mg a pacientes con insuficiencia hepática.
Psiquiátrico
Se han informado juegos patológicos, aumento de la libido e hipersexualidad en pacientes tratados con agonistas de la dopamina, incluida la cabergolina. Esto ha sido generalmente reversible con la reducción de la dosis o la interrupción del tratamiento (Ver Datos de vigilancia posteriores a la comercialización ).
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
Se realizaron estudios de carcinogenicidad en ratones y ratas con cabergolina administrada por sonda en dosis de hasta 0,98 mg/kg/día y 0,32 mg/kg/día, respectivamente. Estas dosis son 7 veces y 4 veces la dosis máxima recomendada en humanos calculada sobre la base del área de superficie corporal usando mg/m2/semana totales en roedores y mg/m2/semana para un ser humano de 50 kg.
Hubo un ligero aumento en la incidencia de leiomiomas cervicales y uterinos y leiomiosarcomas uterinos en ratones. En ratas, hubo un ligero aumento de tumores malignos de cuello uterino y útero y adenomas de células intersticiales. La aparición de tumores en roedores hembra puede estar relacionada con la supresión prolongada de la secreción de prolactina porque la prolactina es necesaria en roedores para el mantenimiento del cuerpo lúteo. En ausencia de prolactina, la relación estrógeno/progesterona aumenta, lo que aumenta el riesgo de tumores uterinos. En roedores machos, la disminución de los niveles de prolactina sérica se asoció con un aumento de la hormona luteinizante sérica, que se cree que es un efecto compensatorio para mantener la síntesis de esteroides testiculares. Dado que se cree que estos mecanismos hormonales son específicos de la especie, se desconoce la relevancia de estos tumores para los seres humanos.
El potencial mutagénico de la cabergolina se evaluó y resultó negativo en una batería de pruebas in vitro. Estas pruebas incluyeron la prueba de mutación bacteriana (Ames) con Salmonella typhimurium, el ensayo de mutación genética con células de hámster chino Schizosaccharomyces pombe P1 y V79, daño y reparación del ADN en Saccharomyces cerevisiae D4 y aberraciones cromosómicas en linfocitos humanos. La cabergolina también fue negativa en la prueba de micronúcleos de médula ósea en el ratón.
En ratas hembra, una dosis diaria de 0,003 mg/kg durante 2 semanas antes del apareamiento y durante todo el período de apareamiento inhibió la concepción. Esta dosis representa aproximadamente 1/28 de la dosis humana máxima recomendada calculada sobre la base del área de superficie corporal utilizando mg/m2/semana totales en ratas y mg/m2/semana para un ser humano de 50 kg.
El embarazo
Efectos teratogénicos - Categoría B
Se han realizado estudios de reproducción con cabergolina en ratones, ratas y conejos administrados por sonda. (Los múltiplos de la dosis humana máxima recomendada en esta sección se calculan sobre la base del área de superficie corporal utilizando mg/m2/semana totales para animales y mg/m2/semana para un ser humano de 50 kg).
Hubo efectos maternotóxicos pero no efectos teratogénicos en ratones que recibieron cabergolina en dosis de hasta 8 mg/kg/día (aproximadamente 55 veces la dosis máxima recomendada en humanos) durante el período de organogénesis.
Una dosis de 0,012 mg/kg/día (aproximadamente 1/7 de la dosis máxima recomendada en humanos) durante el período de organogénesis en ratas provocó un aumento de las pérdidas embriofetales postimplantación. Estas pérdidas podrían deberse a las propiedades inhibidoras de prolactina de la cabergolina en ratas. A dosis diarias de 0,5 mg/kg/día (aproximadamente 19 veces la dosis máxima recomendada en humanos) durante el período de organogénesis en el conejo, la cabergolina provocó maternotoxicidad caracterizada por pérdida de peso corporal y disminución del consumo de alimento. Dosis de 4 mg/kg/día (aproximadamente 150 veces la dosis máxima recomendada en humanos) durante el período de organogénesis en el conejo provocó un aumento de la aparición de diversas malformaciones. Sin embargo, en otro estudio en conejos, no se observaron malformaciones relacionadas con el tratamiento ni embriofetotoxicidad con dosis de hasta 8 mg/kg/día (aproximadamente 300 veces la dosis máxima recomendada en humanos).
En ratas, dosis superiores a 0,003 mg/kg/día (aproximadamente 1/28 de la dosis máxima recomendada en humanos) desde 6 días antes del parto y durante todo el período de lactancia inhibieron el crecimiento y provocaron la muerte de las crías debido a la disminución de la secreción de leche.
Sin embargo, no existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre predicen la respuesta humana, este medicamento debe usarse durante el embarazo solo si es claramente necesario.
Madres lactantes
No se sabe si este fármaco se excreta en la leche humana. Debido a que muchos medicamentos se excretan en la leche humana y debido al potencial de reacciones adversas graves en los lactantes debido a la cabergolina, se debe tomar la decisión de suspender la lactancia o suspender el medicamento, teniendo en cuenta la importancia del medicamento para la madre. No se recomienda el uso de DOSTINEX para la inhibición o supresión de la lactancia fisiológica (ver PRECAUCIONES sección).
La acción reductora de la prolactina de la cabergolina sugiere que interferirá con la lactancia. Debido a esta interferencia con la lactancia, DOSTINEX 0.5 mg no debe administrarse a mujeres posparto que están amamantando o planean amamantar.
Uso pediátrico
No se ha establecido la seguridad y eficacia de DOSTINEX 0,5 mg en pacientes pediátricos.
Uso geriátrico
Los estudios clínicos de DOSTINEX 0,5 mg no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los pacientes más jóvenes. Otra experiencia clínica informada no ha identificado diferencias en las respuestas entre los ancianos y los pacientes más jóvenes. En general, la selección de la dosis para un paciente de edad avanzada debe ser cautelosa, generalmente comenzando en el extremo inferior del rango de dosificación, lo que refleja la mayor frecuencia de disminución de la función hepática, renal o cardíaca, y de enfermedades concomitantes u otra terapia con medicamentos.
SOBREDOSIS
Se puede esperar que la sobredosis produzca congestión nasal, síncope o alucinaciones. Si es necesario, se deben tomar medidas para mantener la presión arterial.
CONTRAINDICACIONES
DOSTINEX Tabletas está contraindicado en pacientes con:
- Hipertensión no controlada o hipersensibilidad conocida a los derivados del cornezuelo.
- Antecedentes de trastornos valvulares cardíacos, según lo sugerido por la evidencia anatómica de valvulopatía de cualquier válvula, determinada por la evaluación previa al tratamiento, incluida la demostración ecocardiográfica de engrosamiento de la valva de la válvula, restricción de la válvula o restricción-estenosis de la válvula mixta. (Ver ADVERTENCIAS )
- Antecedentes de trastornos fibróticos pulmonares, pericárdicos o retroperitoneales. (Ver ADVERTENCIAS )
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
La secreción de prolactina por la hipófisis anterior está principalmente bajo control inhibitorio hipotalámico, probablemente ejercido a través de la liberación de dopamina por las neuronas tuberoinfundibulares. La cabergolina es un agonista de los receptores de dopamina de acción prolongada con una gran afinidad por los receptores D2. Los resultados de los estudios in vitro demuestran que la cabergolina ejerce un efecto inhibidor directo sobre la secreción de prolactina por parte de los lactotrofos hipofisarios de rata. La cabergolina disminuyó los niveles de prolactina sérica en ratas reserpinizadas. Los estudios de unión a receptores indican que la cabergolina tiene baja afinidad por los receptores dopaminérgicos D1, α1 y α2 adrenérgicos y 5-HT1 y 5-HT2 de serotonina.
Estudios clínicos
La eficacia reductora de la prolactina de DOSTINEX 0,25 mg se demostró en mujeres hiperprolactinémicas en dos estudios comparativos aleatorizados, doble ciego, uno con placebo y el otro con bromocriptina. En el estudio controlado con placebo (placebo n=20; cabergolina n=168), DOSTINEX 0.5 mg produjo una disminución relacionada con la dosis en los niveles de prolactina sérica con prolactina normalizada luego de 4 semanas de tratamiento en 29%, 76%, 74% y 95 % de los pacientes que recibieron 0,125, 0,5, 0,75 y 1,0 mg dos veces por semana, respectivamente.
En el período doble ciego de 8 semanas del ensayo comparativo con bromocriptina (cabergolina n=223; bromocriptina n=236 en el análisis por intención de tratar), la prolactina se normalizó en el 77 % de los pacientes tratados con DOSTINEX a 0,5 mg. dos veces por semana en comparación con el 59 % de los tratados con bromocriptina a 2,5 mg dos veces al día. La restauración de la menstruación ocurrió en el 77% de las mujeres tratadas con DOSTINEX, en comparación con el 70% de las tratadas con bromocriptina. Entre los pacientes con galactorrea, este síntoma desapareció en el 73 % de los tratados con DOSTINEX en comparación con el 56 % de los tratados con bromocriptina.
Farmacocinética
Absorción
Después de dosis orales únicas de 0,5 mg a 1,5 mg administradas a 12 voluntarios adultos sanos, se observaron niveles plasmáticos máximos medios de 30 a 70 picogramos (pg)/mL de cabergolina dentro de las 2 a 3 horas. En el rango de dosis de 0,5 a 7 mg, los niveles plasmáticos de cabergolina parecieron ser proporcionales a la dosis en 12 voluntarios adultos sanos y nueve pacientes parkinsonianos adultos. Un estudio de dosis repetidas en 12 voluntarios sanos sugiere que se espera que los niveles de estado estacionario después de un programa de dosificación una vez por semana sean dos o tres veces más altos que después de una dosis única. Se desconoce la biodisponibilidad absoluta de la cabergolina. Una fracción significativa de la dosis administrada sufre un efecto de primer paso. La vida media de eliminación de cabergolina estimada a partir de datos urinarios de 12 sujetos sanos osciló entre 63 y 69 horas. El efecto reductor prolongado de la prolactina de la cabergolina puede estar relacionado con su eliminación lenta y su vida media prolongada.
Distribución
En animales, en base a la radiactividad total, la cabergolina (y/o sus metabolitos) ha mostrado una amplia distribución tisular. La radiactividad en la hipófisis excedió la del plasma en más de 100 veces y se eliminó con una vida media de aproximadamente 60 horas. Este hallazgo es consistente con el efecto reductor de prolactina de larga duración del fármaco. Los estudios de autorradiografía de cuerpo entero en ratas preñadas no mostraron captación fetal pero sí altos niveles en la pared uterina. La radiactividad significativa (principal más metabolitos) detectada en la leche de ratas lactantes sugiere un potencial de exposición para los lactantes. El fármaco se distribuye ampliamente por todo el cuerpo. La cabergolina se une moderadamente (40% a 42%) a las proteínas plasmáticas humanas de manera independiente de la concentración. Es poco probable que la dosificación concomitante de fármacos altamente ligados a proteínas afecte su disposición.
Metabolismo
Tanto en animales como en humanos, la cabergolina se metaboliza ampliamente, predominantemente a través de la hidrólisis del enlace acilurea o la fracción urea. El metabolismo mediado por el citocromo P-450 parece ser mínimo. La cabergolina no provoca inducción y/o inhibición enzimática en la rata. La hidrólisis de la fracción acilurea o urea suprime el efecto reductor de la prolactina de la cabergolina, y los principales metabolitos identificados hasta el momento no contribuyen al efecto terapéutico.
Excreción
Después de la administración oral de cabergolina radiactiva a cinco voluntarios sanos, aproximadamente el 22 % y el 60 % de la dosis se excretó en 20 días en la orina y las heces, respectivamente. Menos del 4% de la dosis se excretó sin cambios en la orina. Las depuraciones no renales y renales de cabergolina son de aproximadamente 3,2 l/min y 0,08 l/min, respectivamente. La excreción urinaria en pacientes hiperprolactinémicos fue similar.
Poblaciones Especiales
Insuficiencia renal
La farmacocinética de la cabergolina no se alteró en 12 pacientes con insuficiencia renal de moderada a grave según lo evaluado por el aclaramiento de creatinina.
insuficiencia hepática
En 12 pacientes con disfunción hepática de leve a moderada (puntuación de Child-Pugh ≤ 10), no se observó ningún efecto sobre la Cmax media de cabergolina o el área bajo la curva de concentración plasmática (AUC). Sin embargo, los pacientes con insuficiencia grave (puntuación de Child-Pugh > 10) muestran un aumento sustancial en la media de Cmax y AUC de cabergolina y, por lo tanto, requieren precaución.
Anciano
No se ha estudiado el efecto de la edad sobre la farmacocinética de la cabergolina.
Interacción Alimento-Fármaco
En 12 voluntarios adultos sanos, la comida no alteró la cinética de la cabergolina.
Farmacodinámica
Se ha documentado la respuesta a la dosis con inhibición de la prolactina plasmática, el inicio del efecto máximo y la duración del efecto después de dosis únicas de cabergolina en voluntarios sanos (0,05 a 1,5 mg) y pacientes hiperprolactinémicos (0,3 a 1 mg). En voluntarios, la inhibición de la prolactina fue evidente con dosis > 0,2 mg, mientras que las dosis ≥ 0,5 mg causaron una supresión máxima en la mayoría de los sujetos. Las dosis más altas producen supresión de la prolactina en una mayor proporción de sujetos y con un inicio más temprano y una duración más prolongada de la acción. En 12 voluntarios sanos, las dosis de 0,5, 1 y 1,5 mg dieron como resultado una inhibición completa de la prolactina, con un efecto máximo dentro de las 3 horas en el 92 % al 100 % de los sujetos después de las dosis de 1 y 1,5 mg en comparación con el 50 % de los sujetos después de la dosis de 0,5 mg. dosis de mg.
En pacientes hiperprolactinémicos (N=51), la disminución máxima de prolactina después de una dosis única de 0,6 mg de cabergolina fue comparable a la de 2,5 mg de bromocriptina; sin embargo, la duración del efecto fue notablemente más prolongada (14 días frente a 24 horas). El tiempo hasta el efecto máximo fue más corto para la bromocriptina que para la cabergolina (6 horas frente a 48 horas).
En 72 voluntarios sanos, dosis únicas o múltiples (hasta 2 mg) de cabergolina dieron como resultado una inhibición selectiva de la prolactina sin efecto aparente sobre otras hormonas de la pituitaria anterior (GH, FSH, LH, ACTH y TSH) o cortisol.
INFORMACIÓN DEL PACIENTE
Se debe instruir a los pacientes para que notifiquen a su médico si sospechan que están embarazadas, quedan embarazadas o tienen la intención de quedar embarazadas durante la terapia. Se debe realizar una prueba de embarazo si existe alguna sospecha de embarazo y se debe discutir con su médico la continuación del tratamiento.
Los pacientes deben informar a su médico si desarrollan dificultad para respirar, tos persistente, dificultad para respirar al acostarse o hinchazón en las extremidades.