Protonix 20mg, 40mg Pantoprazole Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Protonix 40mg y cómo se usa?
Protonix 40 mg es un medicamento recetado que se usa para tratar los síntomas de la esofagitis causada por el ácido estomacal debido a la enfermedad por reflujo gastroesofágico o la ERGE y el síndrome de Zollinger-Ellison u otras afecciones que causan un exceso de ácido estomacal. Protonix 40 mg se puede usar solo o con otros medicamentos.
Protonix pertenece a una clase de medicamentos llamados inhibidores de la bomba de protones (IBP).
No se sabe si Protonix es seguro y eficaz en niños menores de 5 años.
¿Cuáles son los posibles efectos secundarios de Protonix?
Protonix puede causar efectos secundarios graves, que incluyen:
- dolor de estómago severo,
- diarrea acuosa o con sangre,
- dolor repentino o dificultad para mover la cadera, la muñeca o la espalda,
- hematomas o hinchazón en el lugar de la inyección,
- orinar poco o nada,
- sangre en la orina,
- hinchazón,
- rápido aumento de peso,
- mareo,
- frecuencia cardíaca rápida o irregular,
- temblores (sacudidas) o movimientos bruscos,
- sintiéndose nervioso,
- calambres musculares o espasmos en las manos o los pies,
- tos,
- sensación de asfixia,
- dolor en las articulaciones, y
- erupción cutánea en las mejillas o los brazos que empeora por la noche
Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Protonix incluyen:
- dolor de cabeza,
- mareo,
- dolor de estómago,
- gas,
- náuseas,
- vómitos,
- Diarrea,
- dolor en las articulaciones,
- fiebre,
- sarpullido, y
- Síntomas de resfriado (más común en niños)
Informe a su médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Prometrium. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
DESCRIPCIÓN
El ingrediente activo de PROTONIX (pantoprazol sódico) para suspensión oral de liberación retardada y PROTONIX (pantoprazol sódico) tabletas de liberación retardada, un IBP, es un bencimidazol sustituido, 5-(difluorometoxi)-2-[[(3,4- dimetoxi-2-piridinil)metil] sulfinil]-1H-bencimidazol sesquihidrato, un compuesto que inhibe la secreción de ácido gástrico. Su fórmula empírica es C16H14F2N3NaO4S x 1,5 H2O, con un peso molecular de 432,4. La fórmula estructural es:
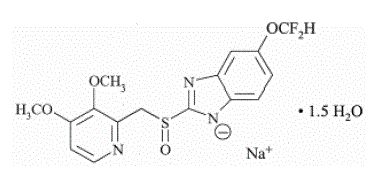
El sesquihidrato de pantoprazol sódico es un polvo cristalino de color blanco a blanquecino y es racémico. Pantoprazol tiene propiedades débilmente básicas y ácidas. El sesquihidrato de pantoprazol sódico es libremente soluble en agua, muy poco soluble en tampón fosfato a pH 7,4 y prácticamente insoluble en n-hexano.
La estabilidad del compuesto en solución acuosa depende del pH. La tasa de degradación aumenta con la disminución del pH. A temperatura ambiente, la vida media de degradación es de aproximadamente 2,8 horas a pH 5 y de aproximadamente 220 horas a pH 7,8.
PROTONIX 40 mg se suministra como suspensión oral de liberación retardada en paquetes de dosis unitarias, disponible en una concentración (40 mg) de pantoprazol (equivalente a 45,1 mg de pantoprazol sódico) y como tableta de liberación retardada, disponible en dos concentraciones 20 mg de pantoprazol (equivalente a 22,56 mg de pantoprazol sódico) y 40 mg de pantoprazol (equivalente a 45,1 mg de pantoprazol sódico).
Las tabletas de liberación retardada PROTONIX contienen los siguientes ingredientes inactivos: estearato de calcio, crospovidona, hipromelosa, óxido de hierro, manitol, copolímero de ácido metacrílico, polisorbato 80, povidona, propilenglicol, carbonato de sodio, laurilsulfato de sodio, dióxido de titanio y citrato de trietilo. Las tabletas de liberación retardada PROTONIX (40 mg y 20 mg) cumplen con la prueba de disolución 2 de la USP.
PROTONIX 40 mg para suspensión oral de liberación retardada contiene los siguientes ingredientes inactivos: crospovidona, hipromelosa, copolímero de ácido metacrílico, celulosa microcristalina, polisorbato 80, povidona, carbonato de sodio, lauril sulfato de sodio, talco, dióxido de titanio, citrato de trietilo y óxido férrico amarillo.
INDICACIONES
PROTONIX para suspensión oral de liberación retardada y PROTONIX 40 mg comprimidos de liberación retardada están indicados para:
Tratamiento a corto plazo de la esofagitis erosiva asociada con la enfermedad por reflujo gastroesofágico (ERGE)
PROTONIX 20 mg está indicado en adultos y pacientes pediátricos a partir de los cinco años de edad para el tratamiento a corto plazo (hasta 8 semanas) en la cicatrización y el alivio sintomático de la esofagitis erosiva (EE). Para aquellos pacientes adultos que no hayan sanado después de 8 semanas de tratamiento, se puede considerar un curso adicional de 8 semanas de PROTONIX. No se ha establecido la seguridad del tratamiento más allá de las 8 semanas en pacientes pediátricos.
Mantenimiento de la curación de la esofagitis erosiva
PROTONIX está indicado para el mantenimiento de la curación de EE y la reducción de las tasas de recaída de los síntomas de acidez estomacal diurna y nocturna en pacientes adultos con ERGE. Los estudios controlados no se extendieron más allá de 12 meses.
Condiciones hipersecretoras patológicas, incluido el síndrome de Zollinger-Ellison
PROTONIX 20 mg está indicado para el tratamiento a largo plazo de condiciones hipersecretoras patológicas, incluido el síndrome de Zollinger-Ellison (ZE).
DOSIFICACIÓN Y ADMINISTRACIÓN
Programa de dosificación recomendado
PROTONIX se presenta como gránulos de liberación retardada en sobres para la preparación de suspensiones orales o como comprimidos de liberación retardada. Las dosis recomendadas se describen en la Tabla 1.
Instrucciones de administración
Las instrucciones para el método de administración de cada forma de dosificación se presentan en la Tabla 2.
Tome una dosis olvidada tan pronto como sea posible. Si es casi la hora de la próxima dosis, omita la dosis olvidada y tome la siguiente dosis a la hora programada. No tome 2 dosis al mismo tiempo.
Tabletas de liberación retardada PROTONIX
Trague las tabletas de liberación retardada de PROTONIX enteras, con o sin alimentos en el estómago. Para los pacientes que no pueden tragar una tableta de 40 mg, se pueden tomar dos tabletas de 20 mg. La administración concomitante de antiácidos no afecta la absorción de las tabletas de liberación retardada de PROTONIX.
PROTONIX para suspensión oral de liberación retardada
Administre PROTONIX para suspensión oral de liberación retardada aproximadamente 30 minutos antes de una comida por vía oral en jugo de manzana o puré de manzana o sonda nasogástrica en jugo de manzana únicamente. Debido a que el pH adecuado es necesario para la estabilidad, no administre PROTONIX 20 mg para suspensión oral de liberación retardada en líquidos que no sean jugo de manzana o alimentos que no sean compota de manzana.
No divida el paquete de 40 mg de PROTONIX 20 mg para suspensión oral de liberación retardada para crear una dosis de 20 mg para pacientes pediátricos que no pueden tomar la formulación en tableta.
PROTONIX para suspensión oral de liberación retardada - Administración oral en compota de manzana
- Paquete abierto.
- Espolvoree los gránulos en una cucharadita de compota de manzana. NO UTILICE OTROS ALIMENTOS NI TRITURE NI MASTICE LOS GRÁNULOS.
- Tomar dentro de los 10 minutos de la preparación.
- Tome sorbos de agua para asegurarse de que los gránulos lleguen al estómago. Repita los sorbos de agua según sea necesario.
PROTONIX para suspensión oral de liberación retardada - Administración oral en jugo de manzana
- Paquete abierto.
- Vacíe los gránulos en una taza pequeña o una cucharadita que contenga una cucharadita de jugo de manzana.
- Revuelva durante 5 segundos (los gránulos no se disolverán) y trague inmediatamente.
- Para asegurarse de tomar la dosis completa, enjuague el recipiente una o dos veces con jugo de manzana para eliminar los gránulos restantes. Trague inmediatamente.
PROTONIX 40 mg para suspensión oral de liberación retardada: administración por sonda nasogástrica (NG) o sonda de gastrostomía
Para los pacientes que tienen una sonda nasogástrica o una sonda de gastrostomía, PROTONIX 40 mg para suspensión oral de liberación retardada se puede administrar de la siguiente manera:
- Retire el émbolo del cilindro de una jeringa con punta de catéter de 2 onzas (60 ml). Deseche el émbolo.
- Conecte la punta del catéter de la jeringa a un tubo de 16 French (o más grande).
- Sostenga la jeringa unida al tubo lo más alto posible mientras administra PROTONIX para suspensión oral de liberación retardada para evitar que el tubo se doble.
- Vacíe el contenido del paquete en el cilindro de la jeringa.
- Agregue 10 ml (2 cucharaditas) de jugo de manzana y golpee y/o agite suavemente el cilindro de la jeringa para ayudar a enjuagar la jeringa y el tubo. Repita al menos dos veces más usando la misma cantidad de jugo de manzana (10 ml o 2 cucharaditas) cada vez. No deben quedar gránulos en la jeringa.
CÓMO SUMINISTRADO
Formas de dosificación y concentraciones
Tabletas de liberación retardada:
- 40 mg de pantoprazol, tabletas biconvexas ovaladas amarillas impresas con PROTONIX (tinta marrón) en un lado
- 20 mg de pantoprazol, tabletas biconvexas ovaladas amarillas impresas con P20 (tinta marrón) en un lado
Para suspensión oral de liberación retardada:
- 40 mg de pantoprazol, gránulos con recubrimiento entérico de color amarillento pálido a marrón oscuro en un paquete de dosis unitaria
Almacenamiento y manipulación
PROTONIX (pantoprazol sódico) Comprimidos de liberación retardada se suministran como comprimidos amarillos, ovalados, biconvexos, de liberación retardada, impresos con PROTONIX (tinta marrón) en un lado que contienen 40 mg de pantoprazol y están disponibles de la siguiente manera:
CDN 0008-0841-81, botellas de 90
PROTONIX (pantoprazol sódico) Comprimidos de liberación retardada se suministran como comprimidos de liberación retardada biconvexos, ovalados, amarillos, impresos con P20 (tinta marrón) en un lado que contienen 20 mg de pantoprazol y están disponibles de la siguiente manera:
CDN 0008-0843-81, botellas de 90
PROTONIX (pantoprazol sódico) para suspensión oral de liberación retardada se suministra como gránulos con cubierta entérica de color amarillento pálido a marrón oscuro que contienen 40 mg de pantoprazol en un paquete de dosis unitaria y están disponibles de la siguiente manera:
CDN 0008-0844-02, caja de dosis unitaria de 30
Almacenamiento
Almacene PROTONIX 40 mg para suspensión oral de liberación retardada y PROTONIX 20 mg tabletas de liberación retardada a una temperatura de 20 ° a 25 ° C (68 ° a 77 ° F); excursiones permitidas a 15° a 30°C (59° a 86°F) [ver Temperatura ambiente controlada por USP ].
Distribuido por: Wyeth Pharmaceuticals LLC, una subsidiaria de Pfizer Inc., Filadelfia, PA 19101. Revisado: noviembre de 2020
EFECTOS SECUNDARIOS
Las siguientes reacciones adversas graves se describen a continuación y en otras partes del etiquetado:
- Nefritis tubulointersticial aguda [ver ADVERTENCIAS Y PRECAUCIONES ]
- Diarrea asociada a Clostridium difficile [ver ADVERTENCIAS Y PRECAUCIONES ]
- Fractura ósea [ver ADVERTENCIAS Y PRECAUCIONES ]
- Lupus eritematoso cutáneo y sistémico [ver ADVERTENCIAS Y PRECAUCIONES ]
- Deficiencia de cianocobalamina (vitamina B-12) [ver ADVERTENCIAS Y PRECAUCIONES ]
- Hipomagnesemia [ver ADVERTENCIAS Y PRECAUCIONES ]
- Pólipos de las glándulas fúndicas [ver ADVERTENCIAS Y PRECAUCIONES ]
Experiencia en ensayos clínicos
Los perfiles de reacciones adversas para PROTONIX (pantoprazol sódico) para suspensión oral de liberación retardada y PROTONIX (pantoprazol sódico) para tabletas de liberación retardada son similares.
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica clínica.
Adultos
La seguridad en nueve ensayos clínicos aleatorios comparativos en EE. UU. en pacientes con ERGE incluyó a 1473 pacientes con PROTONIX oral (20 mg o 40 mg), 299 pacientes con un antagonista del receptor H2, 46 pacientes con otro IBP y 82 pacientes con placebo. Las reacciones adversas más frecuentes se enumeran en la Tabla 3.
Las reacciones adversas adicionales que se informaron para PROTONIX 20 mg en ensayos clínicos con una frecuencia de ≤2% se enumeran a continuación por sistema corporal:
Cuerpo como un todo: reacción alérgica, pirexia, reacción de fotosensibilidad, edema facial
Gastrointestinal: estreñimiento, boca seca, hepatitis
hematológico: leucopenia, trombocitopenia
Metabólico/Nutricional: CK elevada (creatina quinasa), edema generalizado, triglicéridos elevados, enzimas hepáticas elevadas
Musculoesquelético: mialgia
Nervioso: depresión, vértigo
Piel y apéndices: urticaria, erupción, prurito
Sentidos especiales: visión borrosa
Pacientes pediátricos
La seguridad de PROTONIX en el tratamiento de la EE asociada con la ERGE se evaluó en pacientes pediátricos de 1 a 16 años de edad en tres ensayos clínicos. Los ensayos de seguridad incluyeron pacientes pediátricos con EE; sin embargo, dado que la EE es poco común en la población pediátrica, también se evaluaron 249 pacientes pediátricos con ERGE sintomática o comprobada endoscópicamente. Todas las reacciones adversas en adultos a PROTONIX 20 mg se consideran relevantes para pacientes pediátricos. En pacientes de 1 a 16 años de edad, las reacciones adversas notificadas con mayor frecuencia (>4 %) incluyen: URI, dolor de cabeza, fiebre, diarrea, vómitos, erupción cutánea y dolor abdominal.
Para obtener información de seguridad en pacientes menores de 1 año de edad, consulte Uso en poblaciones específicas .
Las reacciones adversas adicionales que se informaron para PROTONIX 20 mg en pacientes pediátricos en ensayos clínicos con una frecuencia de ≤4 % se enumeran a continuación por sistema corporal:
Cuerpo como un todo: reacción alérgica, edema facial
Gastrointestinal: estreñimiento, flatulencia, náuseas
Metabólico/Nutricional: triglicéridos elevados, enzimas hepáticas elevadas, CK elevada (creatina quinasa)
Musculoesquelético: artralgia, mialgia
Nervioso: mareo, vértigo
Piel y apéndices: urticaria
Las siguientes reacciones adversas observadas en adultos en ensayos clínicos no se informaron en pacientes pediátricos en ensayos clínicos, pero se consideran relevantes para pacientes pediátricos: reacción de fotosensibilidad, sequedad de boca, hepatitis, trombocitopenia, edema generalizado, depresión, prurito, leucopenia y visión borrosa .
Síndrome de Zollinger-Ellison (ZE)
En estudios clínicos del síndrome ZE, las reacciones adversas notificadas en 35 pacientes que tomaron PROTONIX de 80 mg/día a 240 mg/día durante un máximo de 2 años fueron similares a las notificadas en pacientes adultos con ERGE.
Experiencia posterior a la comercialización
Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de PROTONIX. Debido a que estas reacciones son informadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Estas reacciones adversas se enumeran a continuación por sistema corporal:
Desórdenes gastrointestinales: pólipos de las glándulas fúndicas
Trastornos generales y condiciones de administración: astenia, fatiga, malestar
hematológico: pancitopenia, agranulocitosis
Trastornos hepatobiliares: daño hepatocelular que conduce a ictericia e insuficiencia hepática
Trastornos del sistema inmunológico: anafilaxia (incluyendo shock anafiláctico), lupus eritematoso sistémico
Infecciones e infestaciones: Diarrea asociada a Clostridium difficile
Investigaciones: cambios de peso
Trastornos del metabolismo y nutricionales: hiponatremia, hipomagnesemia
Trastornos musculoesqueléticos: rabdomiólisis, fractura ósea
Nervioso: ageusia, disgeusia
Desórdenes psiquiátricos: alucinaciones, confusión, insomnio, somnolencia
Trastornos renales y urinarios: nefritis tubulointersticial aguda
Trastornos de la piel y del tejido subcutáneo: reacciones dermatológicas graves (algunas fatales), que incluyen eritema multiforme, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica (TEN, algunas fatales), angioedema (edema de Quincke) y lupus eritematoso cutáneo
INTERACCIONES CON LA DROGAS
La Tabla 4 incluye medicamentos con interacciones farmacológicas clínicamente importantes e interacción con diagnósticos cuando se administran concomitantemente con PROTONIX e instrucciones para prevenirlos o manejarlos.
Consulte la etiqueta de los medicamentos utilizados de forma concomitante para obtener más información sobre las interacciones con los IBP.
ADVERTENCIAS
Incluido como parte de la PRECAUCIONES sección.
PRECAUCIONES
Presencia de malignidad gástrica
En adultos, la respuesta sintomática al tratamiento con PROTONIX no excluye la presencia de cáncer gástrico. Considere un seguimiento adicional y pruebas de diagnóstico en pacientes adultos que tienen una respuesta subóptima o una recaída sintomática temprana después de completar el tratamiento con un IBP. En pacientes mayores, también considere una endoscopia.
Nefritis tubulointersticial aguda
Se ha observado nefritis tubulointersticial aguda (TIN) en pacientes que toman IBP y puede ocurrir en cualquier momento durante la terapia con IBP. Los pacientes pueden presentar diversos signos y síntomas, desde reacciones de hipersensibilidad sintomática hasta síntomas no específicos de disminución de la función renal (p. ej., malestar general, náuseas, anorexia). En las series de casos notificadas, algunos pacientes fueron diagnosticados mediante biopsia y en ausencia de manifestaciones extrarrenales (p. ej., fiebre, exantema o artralgia). Suspender PROTONIX 20 mg y evaluar a los pacientes con sospecha de TIN aguda [ver CONTRAINDICACIONES ].
Diarrea asociada a Clostridium difficile
Los estudios observacionales publicados sugieren que la terapia con IBP como PROTONIX 20 mg puede estar asociada con un mayor riesgo de diarrea asociada a Clostridium difficile, especialmente en pacientes hospitalizados. Este diagnóstico debe considerarse para la diarrea que no mejora [ver REACCIONES ADVERSAS ].
Los pacientes deben usar la dosis más baja y la duración más corta de la terapia con IBP apropiada para la afección que se está tratando.
Fractura de hueso
Varios estudios observacionales publicados sugieren que la terapia con IBP puede estar asociada con un mayor riesgo de fracturas de cadera, muñeca o columna relacionadas con la osteoporosis. El riesgo de fractura aumentó en pacientes que recibieron dosis altas, definidas como múltiples dosis diarias, y terapia con IBP a largo plazo (un año o más). Los pacientes deben usar la dosis más baja y la duración más corta de la terapia con IBP apropiada para la afección que se está tratando. Los pacientes con riesgo de fracturas relacionadas con la osteoporosis deben ser tratados de acuerdo con las pautas de tratamiento establecidas [ver DOSIFICACIÓN Y ADMINISTRACIÓN , REACCIONES ADVERSAS ].
Lupus Eritematoso Cutáneo Y Sistémico
Se han notificado casos de lupus eritematoso cutáneo (CLE) y lupus eritematoso sistémico (LES) en pacientes que toman IBP, incluido pantoprazol sódico. Estos eventos han ocurrido como un nuevo inicio y una exacerbación de la enfermedad autoinmune existente. La mayoría de los casos de lupus eritematoso inducido por IBP fueron LEC.
La forma más común de LEC notificada en pacientes tratados con IBP fue la LEC subaguda (SCLE) y se produjo semanas o años después de la terapia farmacológica continua en pacientes que iban desde bebés hasta ancianos. En general, los hallazgos histológicos se observaron sin afectación de órganos.
El lupus eritematoso sistémico (LES) se notifica con menos frecuencia que el LEC en pacientes que reciben IBP. El LES asociado con IBP suele ser más leve que el LES no inducido por fármacos. El inicio del LES generalmente ocurre dentro de los días o años después de iniciar el tratamiento, principalmente en pacientes que van desde adultos jóvenes hasta ancianos. La mayoría de los pacientes presentaron erupción cutánea; sin embargo, también se informaron artralgia y citopenia.
Evitar la administración de IBP por más tiempo del indicado médicamente. Si se observan signos o síntomas compatibles con CLE o SLE en pacientes que reciben PROTONIX 40 mg, suspenda el medicamento y remita al paciente al especialista adecuado para su evaluación. La mayoría de los pacientes mejoran con la interrupción del PPI solo en 4 a 12 semanas. Las pruebas serológicas (p. ej., ANA) pueden ser positivas y los resultados elevados de las pruebas serológicas pueden tardar más en resolverse que las manifestaciones clínicas.
Deficiencia de cianocobalamina (vitamina B-12)
En general, el tratamiento diario con medicamentos supresores de ácido durante un período prolongado (p. ej., más de 3 años) puede provocar una malabsorción de cianocobalamina (vitamina B-12) causada por hipoclorhidria o aclorhidria. Se han informado en la literatura informes raros de deficiencia de cianocobalamina que ocurren con la terapia supresora de ácido. Este diagnóstico debe considerarse si se observan síntomas clínicos compatibles con la deficiencia de cianocobalamina.
Hipomagnesemia
Rara vez se ha informado hipomagnesemia, sintomática y asintomática, en pacientes tratados con IBP durante al menos tres meses, y en la mayoría de los casos después de un año de terapia. Los eventos adversos graves incluyen tetania, arritmias y convulsiones. En la mayoría de los pacientes, el tratamiento de la hipomagnesemia requirió la reposición de magnesio y la interrupción del PPI.
Para los pacientes que se espera que reciban un tratamiento prolongado o que toman IBP con medicamentos como digoxina o medicamentos que pueden causar hipomagnesemia (p. ej., diuréticos), los profesionales de la salud pueden considerar monitorear los niveles de magnesio antes de iniciar el tratamiento con IBP y periódicamente [ver REACCIONES ADVERSAS ].
Tumorigenicidad
Debido a la naturaleza crónica de la ERGE, puede existir la posibilidad de una administración prolongada de PROTONIX. En estudios a largo plazo con roedores, el pantoprazol fue cancerígeno y causó tipos raros de tumores gastrointestinales. Se desconoce la relevancia de estos hallazgos para el desarrollo de tumores en humanos [ver Toxicología no clínica ].
Pólipos de las glándulas fúndicas
El uso de PPI se asocia con un mayor riesgo de pólipos en las glándulas fúndicas que aumenta con el uso a largo plazo, especialmente más allá de un año. La mayoría de los usuarios de PPI que desarrollaron pólipos en las glándulas fúndicas eran asintomáticos y los pólipos en las glándulas fúndicas se identificaron incidentalmente en la endoscopia. Use la duración más corta de la terapia con PPI apropiada para la afección que se está tratando.
Interferencia con investigaciones de tumores neuroendocrinos
Los niveles séricos de cromogranina A (CgA) aumentan como consecuencia de la disminución de la acidez gástrica inducida por fármacos. El aumento del nivel de CgA puede causar resultados falsos positivos en las investigaciones de diagnóstico de tumores neuroendocrinos. Los proveedores de atención médica deben suspender temporalmente el tratamiento con PROTONIX 20 mg al menos 14 días antes de evaluar los niveles de CgA y considerar repetir la prueba si los niveles iniciales de CgA son altos. Si se realizan pruebas en serie (p. ej., para el control), se debe usar el mismo laboratorio comercial para las pruebas, ya que los rangos de referencia entre las pruebas pueden variar [ver FARMACOLOGÍA CLÍNICA ].
Interferencia con la prueba de orina para THC
Ha habido informes de pruebas de detección de tetrahidrocannabinol (THC) en orina con resultados positivos falsos en pacientes que reciben IBP, incluido PROTONIX [ver INTERACCIONES CON LA DROGAS ].
Uso concomitante de PROTONIX con metotrexato
La literatura sugiere que el uso concomitante de PPI con metotrexato (principalmente en dosis altas; consulte la información de prescripción de metotrexato) puede elevar y prolongar los niveles séricos de metotrexato y/o su metabolito, lo que posiblemente provoque toxicidad por metotrexato. En la administración de dosis altas de metotrexato, en algunos pacientes se puede considerar la suspensión temporal del IBP [ver INTERACCIONES CON LA DROGAS ].
Información de asesoramiento para pacientes
Aconseje al paciente que lea la etiqueta del paciente aprobada por la FDA ( Guía del medicamento e instrucciones de uso ).
Malignidad gástrica
Aconseje a los pacientes que regresen a su proveedor de atención médica si tienen una respuesta subóptima o una recaída sintomática temprana [ver ADVERTENCIAS Y PRECAUCIONES ].
Nefritis tubulointersticial aguda
Aconseje a los pacientes que llamen a su proveedor de atención médica de inmediato si experimentan signos y/o síntomas asociados con la nefritis tubulointersticial aguda [ver CONTRAINDICACIONES , ADVERTENCIAS Y PRECAUCIONES ].
Diarrea asociada a Clostridium difficile
Aconseje a los pacientes que llamen inmediatamente a su proveedor de atención médica si experimentan diarrea que no mejora [ver ADVERTENCIAS Y PRECAUCIONES ].
Fractura de hueso
Aconseje a los pacientes que informen a su proveedor de atención médica sobre cualquier fractura, especialmente de cadera, muñeca o columna [ver ADVERTENCIAS Y PRECAUCIONES ].
Lupus Eritematoso Cutáneo Y Sistémico
Aconseje a los pacientes que llamen de inmediato a su proveedor de atención médica ante cualquier síntoma nuevo o que empeore asociado con el lupus eritematoso cutáneo o sistémico [ver ADVERTENCIAS Y PRECAUCIONES ].
Deficiencia de cianocobalamina (vitamina B-12)
Aconseje a los pacientes que informen a su proveedor de atención médica sobre cualquier síntoma clínico que pueda estar asociado con la deficiencia de cianocobalamina si han estado recibiendo PROTONIX 40 mg durante más de 3 años [ver ADVERTENCIAS Y PRECAUCIONES ].
Hipomagnesemia
Aconseje a los pacientes que informen a su proveedor de atención médica sobre cualquier síntoma clínico que pueda estar asociado con la hipomagnesemia, si han estado recibiendo PROTONIX 40 mg durante al menos 3 meses [ver ADVERTENCIAS Y PRECAUCIONES ].
Interacciones con la drogas
Indique a los pacientes que informen a su proveedor de atención médica sobre cualquier otro medicamento que estén tomando actualmente, incluidos los productos que contienen rilpivirina [ver CONTRAINDICACIONES ] digoxina [ver ADVERTENCIAS Y PRECAUCIONES ] y dosis altas de metotrexato [ver ADVERTENCIAS Y PRECAUCIONES ].
El embarazo
Aconseje a una mujer embarazada sobre el riesgo potencial para el feto. Aconseje a las mujeres con potencial reproductivo que informen a su proveedor de atención médica sobre un embarazo conocido o sospechado [ver Uso en poblaciones específicas ].
Administración
- No parta, triture ni mastique PROTONIX para suspensión oral de liberación retardada y PROTONIX tabletas de liberación retardada de 20 mg.
- El paquete de suspensión oral de PROTONIX es una dosis fija y no se puede dividir para hacer una dosis más pequeña.
- Trague las tabletas de liberación retardada de 20 mg de PROTONIX enteras, con o sin alimentos en el estómago.
- La administración concomitante de antiácidos no afecta la absorción de las tabletas de liberación retardada de PROTONIX.
- Tome PROTONIX 20 mg para suspensión oral de liberación retardada aproximadamente 30 minutos antes de una comida.
- Administre PROTONIX para suspensión oral de liberación retardada en jugo de manzana o puré de manzana, como se describe en las Instrucciones de uso. No administrar en agua, otros líquidos o alimentos.
- Para pacientes con una sonda nasogástrica (NG) o de gastrostomía, PROTONIX para suspensión oral de liberación retardada se puede administrar con jugo de manzana, como se describe en las Instrucciones de uso.
- Tome una dosis olvidada tan pronto como sea posible. Si es casi la hora de la próxima dosis, omita la dosis olvidada y tome la siguiente dosis a la hora programada. No tome 2 dosis al mismo tiempo.
Toxicología no clínica
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
En un estudio de carcinogenicidad de 24 meses, ratas Sprague-Dawley fueron tratadas por vía oral con dosis de pantoprazol de 0,5 a 200 mg/kg/día, alrededor de 0,1 a 40 veces la exposición sobre la base del área de superficie corporal de una persona de 50 kg que recibió una dosis de 40 mg /día. En el fondo gástrico, el tratamiento con 0,5 a 200 mg/kg/día produjo hiperplasia de células tipo enterocromafines (ECL) y tumores de células neuroendocrinas benignas y malignas de manera relacionada con la dosis. En el anteestómago, el tratamiento con 50 y 200 mg/kg/día (alrededor de 10 y 40 veces la dosis humana recomendada sobre la base del área de superficie corporal) produjo papilomas benignos de células escamosas y carcinomas malignos de células escamosas. Los tumores gastrointestinales raros asociados con el tratamiento con pantoprazol incluyeron un adenocarcinoma de duodeno con 50 mg/kg/día y pólipos benignos y adenocarcinomas del fondo gástrico con 200 mg/kg/día. En el hígado, el tratamiento con 0,5 a 200 mg/kg/día produjo aumentos relacionados con la dosis en la incidencia de adenomas y carcinomas hepatocelulares. En la glándula tiroides, el tratamiento con 200 mg/kg/día produjo un aumento de la incidencia de adenomas y carcinomas de células foliculares tanto en ratas macho como hembra.
En un estudio de carcinogenicidad de 24 meses, ratas Fischer 344 fueron tratadas por vía oral con dosis de 5 a 50 mg/kg/día de pantoprazol, aproximadamente de 1 a 10 veces la dosis humana recomendada en función del área de superficie corporal. En el fondo gástrico, el tratamiento con 5 a 50 mg/kg/día produjo hiperplasia de células tipo enterocromafines (ECL) y tumores de células neuroendocrinas benignas y malignas. La selección de dosis para este estudio puede no haber sido adecuada para evaluar exhaustivamente el potencial carcinogénico de pantoprazol.
En un estudio de carcinogenicidad de 24 meses, los ratones B6C3F1 fueron tratados por vía oral con dosis de 5 a 150 mg/kg/día de pantoprazol, de 0,5 a 15 veces la dosis humana recomendada según el área de superficie corporal. En el hígado, el tratamiento con 150 mg/kg/día produjo una mayor incidencia de adenomas y carcinomas hepatocelulares en ratones hembra. El tratamiento con 5 a 150 mg/kg/día también produjo hiperplasia de células ECL gástrico-fúndicas.
Un estudio de carcinogenicidad en ratones transgénicos p53 +/- de 26 semanas no fue positivo.
Pantoprazol fue positivo en los ensayos de aberración cromosómica de linfocitos humanos in vitro, en una de las dos pruebas de micronúcleo de ratón para efectos clastogénicos, y en el ensayo de mutación directa de células ováricas de hámster chino/HGPRT in vitro para efectos mutagénicos. Se observaron resultados equívocos en el ensayo de unión covalente de ADN de hígado de rata in vivo. Pantoprazol fue negativo en el ensayo de mutación de Ames in vitro, el ensayo de síntesis de ADN no programada (UDS) in vitro con hepatocitos de rata, el ensayo de mutación del gen de avance de células de mamífero AS52/GPT in vitro, la prueba de mutación de timidina quinasa in vitro con linfoma de ratón L5178Y y el ensayo de aberración cromosómica de células de médula ósea de rata in vivo.
No hubo efectos sobre la fertilidad o el rendimiento reproductivo cuando se administró pantoprazol en dosis orales de hasta 500 mg/kg/día en ratas macho (98 veces la dosis humana recomendada según el área de superficie corporal) y 450 mg/kg/día en ratas hembra. (88 veces la dosis humana recomendada según el área de superficie corporal).
Uso en poblaciones específicas
El embarazo
Resumen de riesgos
Los datos disponibles de los estudios observacionales publicados no demostraron una asociación de malformaciones mayores u otros resultados adversos del embarazo con pantoprazol.
En estudios de reproducción animal, no se observó evidencia de resultados de desarrollo adversos con pantoprazol. Se han realizado estudios de reproducción en ratas con dosis orales de hasta 450 mg/kg/día (alrededor de 88 veces la dosis humana recomendada) y conejos con dosis orales de hasta 40 mg/kg/día (alrededor de 16 veces la dosis humana recomendada) con administración de pantoprazol durante la organogénesis en animales preñados y no han revelado evidencia de daño al feto debido a pantoprazol en este estudio (ver Datos ).
Se realizó un estudio de toxicidad en el desarrollo pre y posnatal en ratas con criterios de valoración adicionales para evaluar el efecto sobre el desarrollo óseo con pantoprazol sódico. Se administraron dosis orales de pantoprazol de 5, 15 y 30 mg/kg/día (aproximadamente 1, 3 y 6 veces la dosis humana de 40 mg/día) a hembras preñadas desde el día de gestación (GD) 6 hasta el día de lactancia (LD ) 21. Se observaron cambios en la morfología ósea en cachorros expuestos a pantoprazol en el útero y a través de la leche durante el período de lactancia, así como con la administración oral desde el día posnatal (DPN) 4 hasta el DPN 21 [ver Uso en poblaciones específicas ]. No hubo hallazgos relacionados con el fármaco en los animales maternos. Informar a las mujeres embarazadas sobre el riesgo potencial de daño fetal.
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. Todos los embarazos tienen un riesgo de fondo de defecto congénito, pérdida u otros resultados adversos. En la población general de los EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en los embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente.
Datos
Datos humanos
Los datos disponibles de los estudios observacionales publicados no lograron demostrar una asociación de resultados adversos relacionados con el embarazo y el uso de pantoprazol. Las limitaciones metodológicas de estos estudios observacionales no pueden establecer o excluir definitivamente ningún riesgo asociado con el fármaco durante el embarazo. En un estudio prospectivo realizado por la Red Europea de Servicios de Información sobre Teratología, se compararon los resultados de un grupo de 53 mujeres embarazadas a las que se administró una dosis diaria mediana de 40 mg de pantoprazol con un grupo de control de 868 mujeres embarazadas que no tomaron ningún inhibidor de la bomba de protones (IBP). . No hubo diferencia en la tasa de malformaciones mayores entre las mujeres expuestas a IBP y el grupo control, correspondiente a un Riesgo Relativo (RR)=0,55, [95% Intervalo de Confianza (IC) 0,08-3,95]. En un estudio de cohortes retrospectivo basado en la población que abarcó todos los nacidos vivos en Dinamarca desde 1996 hasta 2008, no hubo un aumento significativo de defectos congénitos importantes durante el análisis de la exposición al pantoprazol en el primer trimestre en 549 nacidos vivos. Un metanálisis que comparó 1530 mujeres embarazadas expuestas a IBP en al menos el primer trimestre con 133 410 mujeres embarazadas no expuestas no mostró aumentos significativos en el riesgo de malformaciones congénitas o aborto espontáneo con la exposición a IBP (para malformaciones mayores OR=1.12 ([95% IC 0,86-1,45] y para abortos espontáneos OR=1,29 [IC 95% 0,84-1,97]).
Datos de animales
Se han realizado estudios de reproducción en ratas con dosis orales de pantoprazol de hasta 450 mg/kg/día (alrededor de 88 veces la dosis humana recomendada según el área de superficie corporal) y en conejos con dosis orales de hasta 40 mg/kg/día (alrededor de 16 veces la dosis humana recomendada basada en el área de superficie corporal) con la administración de pantoprazol sódico durante la organogénesis en animales preñados. Los estudios no han revelado evidencia de alteración de la fertilidad o daño al feto debido al pantoprazol.
Se realizó un estudio de toxicidad en el desarrollo pre y posnatal en ratas con criterios de valoración adicionales para evaluar el efecto sobre el desarrollo óseo con pantoprazol sódico. Se administraron dosis orales de pantoprazol de 5, 15 y 30 mg/kg/día (aproximadamente 1, 3 y 6 veces la dosis humana de 40 mg/día sobre la base del área de superficie corporal) a mujeres embarazadas desde el día de la gestación (GD) 6 hasta el día de lactancia (LD) 21. En el día postnatal (PND 4) hasta el PND 21, a los cachorros se les administraron dosis orales de 5, 15 y 30 mg/kg/día (aproximadamente 1, 2,3 y 3,2 veces la exposición ( AUC) en humanos a una dosis de 40 mg). No hubo hallazgos relacionados con el fármaco en los animales maternos. Durante la fase de dosificación previa al destete (PND 4 a 21) de las crías, hubo un aumento de la mortalidad y/o moribunda y una disminución del peso corporal y aumento de peso corporal a 5 mg/kg/día (exposiciones aproximadamente iguales (AUC) en humanos que recibieron los 40 mg de dosis) y dosis más altas. En PND 21, se observaron una disminución de la longitud y el peso medios del fémur y cambios en la masa ósea y la geometría del fémur en las crías con 5 mg/kg/día (exposiciones aproximadamente iguales (AUC) en humanos con la dosis de 40 mg) y dosis más altas. Los hallazgos del fémur incluyeron área total más baja, contenido y densidad de minerales óseos, circunferencia perióstica y endóstica y momento de inercia de la sección transversal. No hubo cambios microscópicos en el fémur distal, la tibia proximal o las articulaciones de la rodilla. Los cambios en los parámetros óseos fueron parcialmente reversibles después de un período de recuperación, con hallazgos en el PND 70 limitados a la densidad mineral ósea cortical/subcortical de la metáfisis inferior del fémur en las crías hembras a 5 mg/kg/día (exposiciones aproximadamente iguales (AUC) en humanos a los 40 mg de dosis) y dosis más altas.
Lactancia
Resumen de riesgos
Se ha detectado pantoprazol en la leche materna de una madre lactante después de una dosis oral única de 40 mg de pantoprazol. No hubo efectos en el lactante amamantado (ver Datos ). No hay datos sobre los efectos del pantoprazol en la producción de leche.

Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de la madre de PROTONIX 40 mg y cualquier efecto adverso potencial en el niño amamantado por pantoprazol o por la afección materna subyacente.
Datos
Se estudió la leche materna de una mujer de 42 años que recibió 40 mg de pantoprazol por vía oral, a los 10 meses posparto, durante 24 horas, para demostrar niveles bajos de pantoprazol presentes en la leche materna. Pantoprazol fue detectable en la leche solo 2 y 4 horas después de la dosis con niveles en la leche de aproximadamente 36 mcg/L y 24 mcg/L, respectivamente. Se observó una relación leche-plasma de 0,022 2 horas después de la administración del fármaco. Pantoprazol no fue detectable (
Uso pediátrico
La seguridad y eficacia de PROTONIX para el tratamiento a corto plazo (hasta ocho semanas) de EE asociado con ERGE se ha establecido en pacientes pediátricos de 1 año a 16 años de edad. No se ha demostrado la eficacia de EE en pacientes menores de 1 año de edad. Además, para pacientes menores de 5 años de edad, no existe una concentración de dosificación adecuada en una formulación adecuada para la edad disponible. Por lo tanto, PROTONIX está indicado para el tratamiento a corto plazo de EE asociado con ERGE en pacientes de 5 años o más. No se ha establecido la seguridad y eficacia de PROTONIX para usos pediátricos que no sean EE.
1 año hasta los 16 años de edad
El uso de PROTONIX en pacientes pediátricos de 1 año a 16 años de edad para el tratamiento a corto plazo (hasta ocho semanas) de EE asociado con ERGE está respaldado por: a) la extrapolación de resultados de estudios adecuados y bien controlados que respaldaron la aprobación de PROTONIX para el tratamiento de EE asociado con ERGE en adultos, y b) estudios de seguridad, eficacia y farmacocinética realizados en pacientes pediátricos [ver Estudios clínicos , FARMACOLOGÍA CLÍNICA ].
La seguridad de PROTONIX 40 mg en el tratamiento de EE asociado con ERGE en pacientes pediátricos de 1 a 16 años de edad se evaluó en tres estudios multicéntricos, aleatorizados, doble ciego, de tratamiento paralelo, en los que participaron 249 pacientes pediátricos, incluidos 8 con EE (4 pacientes edades de 1 año a 5 años y 4 pacientes de 5 años a 11 años). Los niños de 1 año a 5 años con EE diagnosticado endoscópicamente (definido como una puntuación endoscópica de Hetzel-Dent ≥2) fueron tratados una vez al día durante 8 semanas con uno de los dos niveles de dosis de PROTONIX (aproximadamente 0,6 mg/kg o 1,2 mg/kg). ). Los 4 de estos pacientes con EE se curaron (puntuación de Hetzel-Dent de 0 o 1) a las 8 semanas. Debido a que la EE es poco frecuente en la población pediátrica, en estos estudios también se incluyeron predominantemente pacientes pediátricos con ERGE sintomática o comprobada por endoscopia. Los pacientes fueron tratados con un rango de dosis de PROTONIX 20 mg una vez al día durante 8 semanas. Para conocer los resultados de seguridad, consulte REACCIONES ADVERSAS Debido a que estos ensayos pediátricos no tenían placebo, comparador activo o evidencia de una respuesta a la dosis, los ensayos no fueron concluyentes con respecto al beneficio clínico de PROTONIX 20 mg para la ERGE sintomática en la población pediátrica. No se ha establecido la eficacia de PROTONIX 40 mg para el tratamiento de la ERGE sintomática en pacientes pediátricos.
Aunque los datos de los ensayos clínicos respaldan el uso de PROTONIX para el tratamiento a corto plazo de la EE asociada con la ERGE en pacientes pediátricos de 1 año a 5 años, no existe una formulación de dosificación disponible comercialmente adecuada para pacientes menores de 5 años [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
En un análisis farmacocinético poblacional, los valores de aclaramiento en niños de 1 a 5 años con ERGE comprobada por endoscopia tuvieron un valor medio de 2,4 L/h. Después de una dosis equivalente de 1,2 mg/kg (15 mg para ≤12,5 kg y 20 mg para >12,5 a
Recién nacidos hasta menos de un año de edad
No se encontró que PROTONIX 20 mg fuera efectivo en un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, de retiro del tratamiento de 129 pacientes pediátricos de 1 a 11 meses de edad. Los pacientes se inscribieron si tenían ERGE sintomática según el historial médico y no habían respondido a las intervenciones no farmacológicas para la ERGE durante dos semanas. Los pacientes recibieron PROTONIX diariamente durante cuatro semanas en una fase abierta, luego los pacientes fueron aleatorizados en igual proporción para recibir tratamiento de 40 mg de PROTONIX o placebo durante las siguientes cuatro semanas de manera doble ciego. La eficacia se evaluó observando el tiempo desde la aleatorización hasta la interrupción del estudio debido al empeoramiento de los síntomas durante la fase de retiro del tratamiento de cuatro semanas. No hubo diferencia estadísticamente significativa entre PROTONIX 20 mg y placebo en la tasa de interrupción.
En este ensayo, las reacciones adversas que se informaron con mayor frecuencia (diferencia de ≥4 %) en la población tratada en comparación con la población de placebo fueron CK elevada, otitis media, rinitis y laringitis.
En un análisis farmacocinético poblacional, la exposición sistémica fue mayor en pacientes menores de 1 año con ERGE en comparación con los adultos que recibieron una dosis única de 40 mg (la media geométrica del AUC fue un 103 % mayor en los lactantes prematuros y los recién nacidos que recibieron una dosis única de 2,5 mg). de PROTONIX, y un 23 % mayor en lactantes de 1 a 11 meses de edad que reciben una dosis única de aproximadamente 1,2 mg/kg). En estos pacientes, el aclaramiento aparente (CL/F) aumentó con la edad (aclaramiento medio: 0,6 l/h, rango: 0,03 a 3,2 l/h).
Estas dosis dieron como resultado efectos farmacodinámicos sobre el pH gástrico pero no sobre el esofágico. Después de una dosis diaria de 2,5 mg de PROTONIX 20 mg en lactantes prematuros y recién nacidos, hubo un aumento en el pH gástrico medio (de 4,3 al inicio a 5,2 en el estado estacionario) y en el % medio de tiempo que el pH gástrico fue > 4 ( del 60 % al inicio al 80 % en estado estacionario). Después de una dosis diaria de aproximadamente 1,2 mg/kg de PROTONIX 20 mg en lactantes de 1 a 11 meses de edad, hubo un aumento en el pH gástrico medio (de 3,1 al inicio a 4,2 en el estado estacionario) y en el % medio de tiempo que el pH gástrico fue > 4 (del 32 % al inicio al 60 % en el estado estacionario). Sin embargo, no se observaron cambios significativos en el pH intraesofágico medio o el % de tiempo en que el pH esofágico fue
Debido a que PROTONIX 20 mg no demostró ser eficaz en el estudio aleatorizado controlado con placebo en este grupo de edad, no está indicado el uso de PROTONIX para el tratamiento de la ERGE sintomática en lactantes menores de 1 año.
Datos de toxicidad animal
En un estudio de desarrollo pre y posnatal en ratas, a las crías se les administraron dosis orales de pantoprazol de 5, 15 y 30 mg/kg/día (aproximadamente 1, 2,3 y 3,2 veces la exposición (AUC) en niños de 6 a 11 años a una dosis de 40 mg) en el día postnatal (PND 4) hasta el PND 21, además de la exposición de la lactancia a través de la leche. En el PND 21, se observó una disminución de la longitud y el peso medio del fémur y cambios en la masa ósea y la geometría del fémur en la descendencia con 5 mg/kg/día (exposiciones aproximadamente iguales (AUC) en niños de 6 a 11 años con la dosis de 40 mg) y dosis más altas. Los cambios en los parámetros óseos fueron parcialmente reversibles después de un período de recuperación.
En animales neonatos/juveniles (ratas y perros), las toxicidades fueron similares a las observadas en animales adultos, incluidas alteraciones gástricas, disminución de la masa de glóbulos rojos, aumento de lípidos, inducción de enzimas e hipertrofia hepatocelular. Se observó una mayor incidencia de células principales eosinofílicas en ratas adultas y neonatos/juveniles, y atrofia de las células principales en ratas adultas y en perros neonatos/juveniles en la mucosa fúndica de los estómagos en estudios de dosis repetidas. Se notó una recuperación total o parcial de estos efectos en animales de ambos grupos de edad después de un período de recuperación.
Uso geriátrico
En ensayos clínicos estadounidenses a corto plazo, las tasas de curación de EE en los 107 pacientes de edad avanzada (≥65 años) tratados con PROTONIX fueron similares a las encontradas en pacientes menores de 65 años. Las tasas de incidencia de reacciones adversas y anomalías de laboratorio en pacientes de edad 65 años y más fueron similares a los asociados con pacientes menores de 65 años.
SOBREDOSIS
La experiencia en pacientes que toman dosis muy altas de PROTONIX (superiores a 240 mg) es limitada. Los informes espontáneos de sobredosis posteriores a la comercialización generalmente se encuentran dentro del perfil de seguridad conocido de PROTONIX.
Pantoprazol no se elimina por hemodiálisis. En caso de sobredosis, el tratamiento debe ser sintomático y de apoyo.
Las dosis orales únicas de pantoprazol de 709 mg/kg, 798 mg/kg y 887 mg/kg fueron letales para ratones, ratas y perros, respectivamente. Los síntomas de toxicidad aguda fueron hipoactividad, ataxia, sedestación encorvada, separación de las extremidades, posición lateral, segregación, ausencia de reflejo auricular y temblor.
Si ocurre una sobreexposición a PROTONIX 20 mg, llame a su centro de control de envenenamiento al 1-800-222-1222 para obtener información actualizada sobre el manejo de envenenamiento o sobredosis.
CONTRAINDICACIONES
- PROTONIX está contraindicado en pacientes con hipersensibilidad conocida a cualquier componente de la formulación o cualquier benzimidazol sustituido. Las reacciones de hipersensibilidad pueden incluir anafilaxia, shock anafiláctico, angioedema, broncoespasmo, nefritis tubulointersticial aguda y urticaria [ver ADVERTENCIAS Y PRECAUCIONES , REACCIONES ADVERSAS ].
- Los inhibidores de la bomba de protones (IBP), incluido PROTONIX, están contraindicados en pacientes que reciben productos que contienen rilpivirina [ver INTERACCIONES CON LA DROGAS ].
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
Pantoprazol es un IBP que suprime el paso final en la producción de ácido gástrico mediante la unión covalente al sistema enzimático (H+, K+)-ATPasa en la superficie secretora de la célula parietal gástrica. Este efecto conduce a la inhibición de la secreción de ácido gástrico tanto basal como estimulada, independientemente del estímulo. La unión a la (H+, K+)-ATPasa da como resultado una duración del efecto antisecretor que persiste más de 24 horas para todas las dosis probadas (20 mg a 120 mg).
Farmacodinámica
PROTONIX 20 mg para suspensión oral de liberación retardada, 40 mg ha demostrado ser comparable a PROTONIX tabletas de liberación retardada en la supresión de la MAO estimulada por pentagastrina en pacientes (n = 49) con ERGE y antecedentes de EE. En este estudio farmacodinámico cruzado multicéntrico, se comparó una dosis oral de 40 mg de PROTONIX para suspensión oral de liberación retardada administrada en una cucharadita de puré de manzana con una dosis oral de 40 mg de comprimidos de liberación retardada de PROTONIX después de la administración de cada formulación una vez al día durante 7 días. Ambos medicamentos se administraron treinta minutos antes del desayuno. La estimulación con pentagastrina (MAO) se evaluó desde la hora 23 a la 24 en estado estacionario.
Actividad antisecretora
En condiciones de máxima estimulación ácida usando pentagastrina, se produce una disminución dependiente de la dosis en la producción de ácido gástrico después de una dosis única de pantoprazol por vía oral (20-80 mg) o intravenosa (20-120 mg) en sujetos sanos. Pantoprazol administrado una vez al día da como resultado un aumento de la inhibición de la secreción de ácido gástrico. Tras la dosis oral inicial de 40 mg de pantoprazol, se alcanzó una inhibición media del 51 % a las 2,5 horas. Con la dosificación una vez al día durante 7 días, la inhibición media aumentó al 85 %. Pantoprazol suprimió la secreción de ácido en más del 95% en la mitad de los sujetos. La secreción de ácido había vuelto a la normalidad una semana después de la última dosis de pantoprazol; no hubo evidencia de hipersecreción de rebote.
En una serie de estudios de dosis-respuesta, pantoprazol, en dosis orales que oscilaron entre 20 y 120 mg, provocó aumentos relacionados con la dosis en el pH gástrico basal medio y en el porcentaje de tiempo que el pH gástrico fue >3 y >4. El tratamiento con 40 mg de pantoprazol produjo aumentos significativamente mayores en el pH gástrico que la dosis de 20 mg. Las dosis superiores a 40 mg (60, 80, 120 mg) no produjeron aumentos significativos adicionales en el pH gástrico medio. Los efectos del pantoprazol en la mediana del pH de un estudio doble ciego cruzado se muestran en la Tabla 5.
Efectos de la gastrina sérica
Los niveles de gastrina sérica en ayunas se evaluaron en dos estudios doble ciego de curación aguda de EE en los que 682 pacientes con enfermedad por reflujo gastroesofágico (ERGE) recibieron 10, 20 o 40 mg de PROTONIX 20 mg durante un máximo de 8 semanas. A las 4 semanas de tratamiento hubo un aumento en los niveles medios de gastrina del 7 %, 35 % y 72 % sobre los valores previos al tratamiento en los grupos de tratamiento de 10, 20 y 40 mg, respectivamente. Se observó un aumento similar en los niveles de gastrina sérica en la visita de 8 semanas con aumentos medios del 3 %, 26 % y 84 % para los tres grupos de dosis de pantoprazol. Los niveles medianos de gastrina en suero se mantuvieron dentro de los límites normales durante la terapia de mantenimiento con las tabletas de liberación retardada de PROTONIX.
En estudios internacionales a largo plazo en los que participaron más de 800 pacientes, se observó un aumento medio de 2 a 3 veces del nivel de gastrina sérica en ayunas previo al tratamiento en los meses iniciales de tratamiento con pantoprazol en dosis de 40 mg por día durante los estudios de mantenimiento de ERGE y 40 mg o más por día en pacientes con ERGE refractaria. Los niveles de gastrina sérica en ayunas generalmente se mantuvieron en aproximadamente 2 a 3 veces los valores iniciales durante hasta 4 años de seguimiento periódico en ensayos clínicos.
Después de un tratamiento a corto plazo con PROTONIX 20 mg, los niveles elevados de gastrina vuelven a la normalidad en al menos 3 meses.
Efectos de células similares a enterocromafines (ECL)
En 39 pacientes tratados con pantoprazol oral de 40 mg a 240 mg al día (la mayoría recibió de 40 mg a 80 mg) durante un máximo de 5 años, hubo un aumento moderado en la densidad de células ECL, que comenzó después del primer año de uso, que pareció disminuir. meseta después de 4 años.
En un estudio no clínico en ratas Sprague-Dawley, la exposición de por vida (24 meses) a pantoprazol en dosis de 0.5 a 200 mg/kg/día resultó en aumentos relacionados con la dosis en la proliferación de células ECL gástricas y tumores de células neuroendocrinas (NE) gástricas . Los tumores gástricos de células NE en ratas pueden resultar de la elevación crónica de las concentraciones séricas de gastrina. La alta densidad de células ECL en el estómago de la rata hace que esta especie sea muy susceptible a los efectos proliferativos de las concentraciones elevadas de gastrina producidas por los IBP. Sin embargo, no se observaron elevaciones de la gastrina sérica tras la administración de pantoprazol a una dosis de 0,5 mg/kg/día. En un estudio separado, se observó un tumor gástrico de células NE sin cambios concomitantes en la proliferación de células ECL en 1 rata hembra después de 12 meses de dosificación con pantoprazol a 5 mg/kg/día y una recuperación de 9 meses sin la dosis [ver Toxicología no clínica ].
Efectos endocrinos
En un estudio de farmacología clínica, PROTONIX 40 mg administrado una vez al día durante 2 semanas no tuvo efecto sobre los niveles de las siguientes hormonas: cortisol, testosterona, triyodotironina (T3), tiroxina (T4), hormona estimulante de la tiroides (TSH), tironina- proteína de unión, hormona paratiroidea, insulina, glucagón, renina, aldosterona, hormona estimulante del folículo, hormona luteinizante, prolactina y hormona del crecimiento.
En un estudio de 1 año de pacientes con ERGE tratados con PROTONIX 40 mg o 20 mg, no hubo cambios desde el inicio en los niveles generales de T3, T4 y TSH.
Farmacocinética
Los comprimidos de liberación retardada de 40 mg de PROTONIX se preparan como comprimidos con cubierta entérica, de modo que la absorción de pantoprazol comienza solo después de que el comprimido sale del estómago. La concentración sérica máxima (Cmax) y el área bajo la curva de tiempo de concentración sérica (AUC) aumentan de manera proporcional a las dosis orales e intravenosas de 10 mg a 80 mg. Pantoprazol no se acumula y su farmacocinética no se altera con múltiples dosis diarias. Después de la administración oral o intravenosa, la concentración sérica de pantoprazol disminuye biexponencialmente, con una vida media de eliminación terminal de aproximadamente una hora.
En los metabolizadores rápidos con función hepática normal que reciben una dosis oral de la tableta de pantoprazol con cubierta entérica de 40 mg, la concentración máxima (Cmax) es de 2.5 μg/mL; el tiempo para alcanzar la concentración máxima (tmax) es de 2,5 h, y el área total media bajo la curva de concentración plasmática versus tiempo (AUC) es de 4,8 μg·h/ml (rango de 1,4 a 13,3 μg·h/ml). Tras la administración intravenosa de pantoprazol a metabolizadores rápidos, su aclaramiento total es de 7,6 a 14,0 l/h y su volumen aparente de distribución es de 11,0 a 23,6 l.
Se demostró que una dosis oral única de PROTONIX para suspensión oral de liberación retardada, 40 mg, es bioequivalente cuando se administra a sujetos sanos (N = 22) como gránulos rociados sobre una cucharadita de puré de manzana, como gránulos mezclados con jugo de manzana o mezclados con jugo de manzana seguido de la administración a través de una sonda nasogástrica. Los parámetros farmacocinéticos plasmáticos de un estudio cruzado en sujetos sanos se resumen en la Tabla 6.
Absorción
Después de la administración de dosis orales únicas o múltiples de 40 mg de PROTONIX 20 mg comprimidos de liberación retardada, la concentración plasmática máxima de pantoprazol se alcanzó en aproximadamente 2,5 horas y la Cmáx fue de 2,5 μg/ml. El pantoprazol sufre un pequeño metabolismo de primer paso, lo que da como resultado una biodisponibilidad absoluta de aproximadamente el 77 %. La absorción de pantoprazol no se ve afectada por la administración concomitante de antiácidos.
La administración de comprimidos de liberación retardada de PROTONIX con alimentos puede retrasar su absorción hasta 2 horas o más; sin embargo, la Cmax y el grado de absorción de pantoprazol (AUC) no se modifican. Por lo tanto, las tabletas de liberación retardada de 40 mg de PROTONIX se pueden tomar sin tener en cuenta el horario de las comidas.
La administración de gránulos de pantoprazol, 40 mg, con una comida rica en grasas retrasó el tiempo medio hasta la concentración plasmática máxima en 2 horas. Con una comida rica en grasas concomitante, la Cmax y el AUC de los gránulos de pantoprazol, 40 mg, rociados sobre compota de manzana disminuyeron en un 51 % y un 29 %, respectivamente. Por lo tanto, PROTONIX 20 mg para suspensión oral de liberación retardada debe tomarse aproximadamente 30 minutos antes de una comida.
Distribución
El volumen aparente de distribución de pantoprazol es de aproximadamente 11 a 23,6 L, distribuyéndose principalmente en el líquido extracelular. La unión a proteínas séricas de pantoprazol es de alrededor del 98%, principalmente a la albúmina.
Eliminación
Metabolismo
Pantoprazol se metaboliza ampliamente en el hígado a través del sistema del citocromo P450 (CYP). El metabolismo de pantoprazol es independiente de la vía de administración (intravenosa u oral). La vía metabólica principal es la desmetilación, por CYP2C19, con la subsiguiente sulfatación; otras vías metabólicas incluyen la oxidación por CYP3A4. No hay evidencia de que alguno de los metabolitos de pantoprazol tenga una actividad farmacológica significativa.
Excreción
Después de una dosis oral o intravenosa única de pantoprazol marcado con 14C a sujetos sanos metabolizadores normales, aproximadamente el 71 % de la dosis se excretó en la orina y el 18 % se excretó en las heces a través de la excreción biliar. No hubo excreción renal de pantoprazol inalterado.
Poblaciones Específicas
Pacientes geriátricos
Solo se encontraron aumentos leves a moderados en el AUC (43 %) y la Cmax (26 %) de pantoprazol en sujetos de edad avanzada (64 a 76 años de edad) después de la administración oral repetida, en comparación con sujetos más jóvenes [ver Uso en poblaciones específicas ].
Pacientes pediátricos
La farmacocinética de pantoprazol se estudió en niños menores de 16 años en cuatro ensayos clínicos aleatorizados y abiertos en pacientes pediátricos con ERGE presunto/comprobado. Se estudió una formulación de gránulos pediátricos en niños de hasta 5 años de edad, y las tabletas de liberación retardada de 40 mg de PROTONIX se estudiaron en niños mayores de 5 años.
En un análisis farmacocinético poblacional, el aclaramiento total aumentó con el aumento del peso corporal de forma no lineal. El aclaramiento total también aumentó con el aumento de la edad solo en niños menores de 3 años.
Recién nacido hasta los 5 años de edad
[ver Uso en poblaciones específicas ]
Niños y Adolescentes de 6 a 16 Años de Edad
La farmacocinética de las tabletas de liberación retardada de PROTONIX se evaluó en niños de 6 a 16 años con diagnóstico clínico de ERGE. Los parámetros farmacocinéticos después de una dosis oral única de 20 mg o 40 mg de comprimidos de 40 mg de PROTONIX en niños de 6 a 16 años fueron muy variables (el % CV oscila entre el 40 y el 80 %). La media geométrica del AUC estimada a partir del análisis farmacocinético poblacional después de un comprimido de 40 mg de PROTONIX de 40 mg en pacientes pediátricos fue aproximadamente un 39 % y un 10 % mayor, respectivamente, en niños de 6 a 11 y de 12 a 16 años, en comparación con la de los adultos (Tabla 7). .
Pacientes masculinos y femeninos
Hay un aumento modesto en el AUC y la Cmax de pantoprazol en mujeres en comparación con los hombres. Sin embargo, los valores de depuración normalizados por peso son similares en mujeres y hombres.
En pacientes pediátricos de 1 a 16 años de edad, no hubo efectos clínicamente relevantes del sexo sobre la depuración de pantoprazol, como lo demuestra el análisis farmacocinético poblacional.
Pacientes con insuficiencia renal
En pacientes con insuficiencia renal grave, los parámetros farmacocinéticos de pantoprazol fueron similares a los de sujetos sanos.
Pacientes con insuficiencia hepática
En pacientes con insuficiencia hepática de leve a grave (cirrosis Child-Pugh A a C), las concentraciones máximas de pantoprazol aumentaron solo ligeramente (1,5 veces) en relación con los sujetos sanos. Aunque los valores de semivida sérica aumentaron de 7 a 9 horas y los valores de AUC aumentaron de 5 a 7 veces en pacientes con insuficiencia hepática, estos aumentos no fueron mayores que los observados en metabolizadores lentos de CYP2C19, en los que no se justifica un ajuste de dosis. Estos cambios farmacocinéticos en pacientes con insuficiencia hepática dan como resultado una acumulación mínima del fármaco después de la administración de dosis múltiples una vez al día. No se han estudiado dosis superiores a 40 mg/día en pacientes con insuficiencia hepática.
Estudios de interacción farmacológica
Efecto de otras drogas sobre pantoprazol
Pantoprazol se metaboliza principalmente por CYP2C19 y, en menor medida, por CYP 3A4, 2D6 y 2C9. Estudios de interacción farmacológica in vivo con sustratos de CYP2C19 (diazepam [también un sustrato de CYP3A4] y fenitoína [también un inductor de CYP3A4] y clopidogrel), nifedipina, midazolam y claritromicina (sustratos de CYP3A4), metoprolol (un sustrato de CYP2D6), diclofenaco , naproxeno y piroxicam (sustratos de CYP2C9) y teofilina (un sustrato de CYP1A2) en sujetos sanos, la farmacocinética de pantoprazol no se alteró significativamente.
Efecto de pantoprazol en otras drogas
clopidogrel
Clopidogrel se metaboliza a su metabolito activo en parte por CYP2C19. En un estudio clínico cruzado, a 66 sujetos sanos se les administró clopidogrel (dosis de carga de 300 mg seguida de 75 mg por día) solo y con pantoprazol (80 mg al mismo tiempo que clopidogrel) durante 5 días. En el día 5, el AUC medio del metabolito activo de clopidogrel se redujo en aproximadamente un 14 % (la relación media geométrica fue del 86 %, con un IC del 90 % de 79 a 93 %) cuando se administró pantoprazol junto con clopidogrel en comparación con clopidogrel administrado solo. También se midieron los parámetros farmacodinámicos y se demostró que el cambio en la inhibición de la agregación plaquetaria (inducida por ADP 5 μM) se correlacionó con el cambio en la exposición al metabolito activo de clopidogrel. La importancia clínica de este hallazgo no está clara.
Micofenolato de mofetilo (MMF)
La administración de pantoprazol 40 mg dos veces al día durante 4 días y una dosis única de 1000 mg de MMF aproximadamente una hora después de la última dosis de pantoprazol a 12 sujetos sanos en un estudio cruzado dio como resultado una reducción del 57 % en la Cmax y una reducción del 27 % en las AUC de MPA. Los pacientes trasplantados que recibieron aproximadamente 2000 mg por día de MMF (n=12) se compararon con pacientes trasplantados que recibieron aproximadamente la misma dosis de MMF y pantoprazol 40 mg por día (n=21). Hubo una reducción del 78 % en la Cmax y una reducción del 45 % en el AUC de MPA en pacientes que recibieron pantoprazol y MMF [ver INTERACCIONES CON LA DROGAS ].
Otras drogas
Los estudios in vivo también sugieren que pantoprazol no afecta significativamente la cinética de los siguientes fármacos (cisaprida, teofilina, diazepam [y su metabolito activo, desmetildiazepam], fenitoína, metoprolol, nifedipina, carbamazepina, midazolam, claritromicina, diclofenaco, naproxeno, piroxicam, y anticonceptivos orales [levonorgestrel/etinilestradiol]). En otros estudios in vivo, la digoxina, el etanol, la gliburida, la antipirina, la cafeína, el metronidazol y la amoxicilina no tuvieron interacciones clínicamente relevantes con pantoprazol.
Aunque no se han observado interacciones farmacológicas significativas en los estudios clínicos, no se ha estudiado el potencial de interacciones farmacológicas significativas con dosis altas de pantoprazol más de una vez al día en metabolizadores lentos o personas con insuficiencia hepática.
antiácidos
Tampoco hubo interacción con los antiácidos administrados concomitantemente.
farmacogenómica
CYP2C19 muestra un polimorfismo genético conocido debido a su deficiencia en algunas subpoblaciones (p. ej., aproximadamente el 3 % de los caucásicos y afroamericanos y el 17 % al 23 % de los asiáticos son metabolizadores lentos). Aunque estas subpoblaciones de metabolizadores lentos de pantoprazol tienen valores de vida media de eliminación de 3,5 a 10 horas en adultos, todavía tienen una acumulación mínima (23% o menos) con la dosificación una vez al día. Para pacientes adultos que son metabolizadores lentos de CYP2C19, no es necesario ajustar la dosis.
Al igual que los adultos, los pacientes pediátricos que tienen el genotipo de metabolizador lento de CYP2C19 (CYP2C19 *2/*2) exhibieron un aumento de más de 6 veces en el AUC en comparación con los pacientes pediátricos extensos (CYP2C19 *1/*1) e intermedios (CYP2C19 *1). /*x) metabolizadores. Los metabolizadores lentos exhibieron una depuración oral aparente aproximadamente 10 veces menor en comparación con los metabolizadores rápidos.
Para los metabolizadores lentos pediátricos conocidos, se debe considerar una reducción de la dosis.
Estudios clínicos
Los comprimidos de liberación retardada de 20 mg de PROTONIX se utilizaron en los siguientes ensayos clínicos.
Esofagitis erosiva (EE) asociada con enfermedad por reflujo gastroesofágico (ERGE)
Pacientes adultos
Se realizó un estudio multicéntrico, doble ciego, controlado con placebo en EE. UU. de PROTONIX 10 mg, 20 mg o 40 mg una vez al día en 603 pacientes con síntomas de reflujo y EE diagnosticado endoscópicamente de grado 2 o superior (escala de Hetzel-Dent). En este estudio, aproximadamente el 25 % de los pacientes incluidos tenían EE grave de grado 3 y el 10 % tenían EE de grado 4. Los porcentajes de pacientes curados (por protocolo, n = 541) en este estudio se muestran en la Tabla 8.
En este estudio, todos los grupos de tratamiento con PROTONIX tuvieron tasas de curación significativamente mayores que el grupo de placebo. Esto fue cierto independientemente del estado de H. pylori para los grupos de tratamiento de 40 mg y 20 mg de PROTONIX 40 mg. La dosis de 40 mg de PROTONIX 20 mg resultó en tasas de curación significativamente mayores que las encontradas con la dosis de 20 mg o 10 mg.
Una proporción significativamente mayor de pacientes que tomaron PROTONIX 40 mg experimentaron un alivio completo de la acidez estomacal diurna y nocturna y la ausencia de regurgitación, desde el primer día de tratamiento, en comparación con el placebo. Los pacientes que tomaron PROTONIX consumieron significativamente menos tabletas de antiácido por día que los que tomaron placebo.
PROTONIX 40 mg y 20 mg una vez al día también se compararon con nizatidina 150 mg dos veces al día en un estudio doble ciego multicéntrico de EE. UU. de 243 pacientes con síntomas de reflujo y EE de grado 2 o superior diagnosticado por endoscopia. Los porcentajes de pacientes curados (por protocolo, n = 212) se muestran en la Tabla 9.
El tratamiento una vez al día con PROTONIX 40 mg o 20 mg resultó en tasas de curación significativamente superiores a las 4 y 8 semanas en comparación con el tratamiento dos veces al día con 150 mg de nizatidina. Para el grupo de tratamiento de 40 mg, se lograron tasas de curación significativamente mayores en comparación con la nizatidina, independientemente del estado de H. pylori.
Una proporción significativamente mayor de pacientes en los grupos de tratamiento con PROTONIX experimentó un alivio completo de la acidez estomacal nocturna y la regurgitación, comenzando el primer día y de la acidez estomacal diurna el segundo día, en comparación con los que tomaron 150 mg de nizatidina dos veces al día. Los pacientes que tomaron PROTONIX consumieron significativamente menos tabletas de antiácido por día que los que tomaron nizatidina.
Pacientes pediátricos de 5 a 16 años de edad
La eficacia de PROTONIX en el tratamiento de la EE asociada con la ERGE en pacientes pediátricos de 5 a 16 años se extrapola de ensayos adecuados y bien realizados en adultos, ya que se cree que la fisiopatología es la misma. Se estudiaron cuatro pacientes pediátricos con EE diagnosticada por endoscopia en ensayos multicéntricos, aleatorizados, doble ciego y de tratamiento paralelo. Los niños con EE diagnosticado por endoscopia (definido como una puntuación de Hetzel-Dent endoscópica ≥2) fueron tratados una vez al día durante 8 semanas con uno de los dos niveles de dosis de PROTONIX (20 mg o 40 mg). Los 4 pacientes con EE se curaron (puntuación de Hetzel-Dent de 0 o 1) a las 8 semanas.
Mantenimiento a largo plazo de la curación de la esofagitis erosiva
Se realizaron dos ensayos independientes, multicéntricos, aleatorizados, doble ciego, controlados con comparador y de diseño idéntico en pacientes adultos con ERGE con EE curado confirmado por endoscopia para demostrar la eficacia de PROTONIX en el mantenimiento a largo plazo de la curación. Los dos estudios estadounidenses reclutaron a 386 y 404 pacientes, respectivamente, para recibir 10 mg, 20 mg o 40 mg de PROTONIX 40 mg comprimidos de liberación retardada una vez al día o 150 mg de ranitidina dos veces al día. Como se demuestra en la Tabla 10, PROTONIX 40 mg y 20 mg fueron significativamente superiores a la ranitidina en todos los puntos de tiempo con respecto al mantenimiento de la cicatrización. Además, PROTONIX 40 mg fue superior a todos los demás tratamientos estudiados.
PROTONIX 40 mg fue superior a la ranitidina en la reducción del número de episodios de acidez estomacal durante el día y la noche desde el primer hasta el duodécimo mes de tratamiento. PROTONIX 20 mg, administrado una vez al día, también fue eficaz para reducir los episodios de acidez estomacal diurna y nocturna en un ensayo, como se presenta en la Tabla 11.
Condiciones hipersecretoras patológicas, incluido el síndrome de Zollinger-Ellison
En un ensayo abierto multicéntrico de 35 pacientes con afecciones hipersecretoras patológicas, como el síndrome de Zollinger-Ellison, con o sin neoplasia endocrina múltiple tipo I, PROTONIX controló con éxito la secreción de ácido gástrico. Las dosis que oscilaron entre 80 mg diarios y 240 mg diarios mantuvieron la producción de ácido gástrico por debajo de 10 mEq/h en pacientes sin cirugía reductora de ácido previa y por debajo de 5 mEq/h en pacientes con cirugía reductora de ácido previa.
Las dosis se ajustaron inicialmente a las necesidades individuales del paciente y se ajustaron en algunos pacientes en función de la respuesta clínica con el tiempo [ver DOSIFICACIÓN Y ADMINISTRACIÓN ]. PROTONIX 40 mg fue bien tolerado a estos niveles de dosis durante períodos prolongados (más de 2 años en algunos pacientes).
INFORMACIÓN DEL PACIENTE
PROTONIX (pro-TAH-nix)(pantoprazol sódico) tabletas de liberación retardada y PROTONIX (pro-TAH-nix) (pantoprazol sódico) para suspensión oral de liberación retardada
¿Cuál es la información más importante que debo saber sobre PROTONIX?
Debe tomar PROTONIX 20 mg exactamente como se le recetó, en la dosis más baja posible y durante el menor tiempo necesario.
PROTONIX puede ayudar con los síntomas relacionados con el ácido, pero aún podría tener problemas estomacales graves. Hable con su médico.
PROTONIX puede causar efectos secundarios graves, que incluyen:
- Un tipo de problema renal (nefritis tubulointersticial aguda). Algunas personas que toman medicamentos inhibidores de la bomba de protones (IBP), incluido PROTONIX 40 mg, pueden desarrollar un problema renal llamado nefritis tubulointersticial aguda que puede ocurrir en cualquier momento durante el tratamiento con PROTONIX. Llame a su médico de inmediato si tiene una disminución en la cantidad que orina o si tiene sangre en la orina.
- Diarrea causada por una infección (Clostridium difficile) en sus intestinos. Llame a su médico de inmediato si tiene heces acuosas o dolor de estómago que no desaparece. Puede o no tener fiebre.
- Fracturas óseas (cadera, muñeca o columna). Las fracturas de huesos en la cadera, la muñeca o la columna vertebral pueden ocurrir en personas que toman múltiples dosis diarias de medicamentos PPI y durante un período prolongado (un año o más). Informe a su médico si tiene una fractura de hueso, especialmente en la cadera, la muñeca o la columna.
- Ciertos tipos de lupus eritematoso. El lupus eritematoso es un trastorno autoinmune (las células inmunitarias del cuerpo atacan a otras células u órganos del cuerpo). Algunas personas que toman medicamentos PPI, incluido PROTONIX 40 mg, pueden desarrollar ciertos tipos de lupus eritematoso o empeorar el lupus que ya tienen. Llame a su médico de inmediato si tiene un dolor en las articulaciones nuevo o que empeora, o una erupción en las mejillas o los brazos que empeora con el sol.
Hable con su médico acerca de su riesgo de sufrir estos efectos secundarios graves.
PROTONIX puede tener otros efectos secundarios graves. Ver “¿Cuáles son los posibles efectos secundarios de PROTONIX?”
¿Qué es PROTONIX 20mg?
Un medicamento recetado llamado inhibidor de la bomba de protones (IBP) que se usa para reducir la cantidad de ácido en el estómago.
En adultos, PROTONIX se utiliza para:
- hasta 8 semanas para la curación y el alivio de los síntomas del daño relacionado con el ácido en el revestimiento del esófago (llamado esofagitis erosiva o EE). Su médico puede prescribir otras 8 semanas de PROTONIX 20 mg en pacientes cuyo EE no se cura.
- mantener la curación de la EE y ayudar a prevenir el regreso de los síntomas de acidez estomacal causados por la ERGE. No se sabe si PROTONIX 40 mg es seguro y efectivo cuando se usa durante más de 12 meses para este fin.
- el tratamiento a largo plazo de condiciones en las que su estómago produce demasiado ácido. Esto incluye una condición rara llamada Síndrome de Zollinger-Ellison.
En niños de 5 años de edad y mayores, PROTONIX se utiliza para:
- hasta 8 semanas para la curación y el alivio de los síntomas de la EE. No se sabe si PROTONIX 20 mg es seguro si se usa durante más de 8 semanas en niños.
PROTONIX 20 mg no es para uso en niños menores de 5 años. No se sabe si PROTONIX 40 mg es seguro y eficaz en niños para un tratamiento distinto de EE.
No tome PROTONIX 20 mg si usted es:
- alérgico al pantoprazol sódico, a cualquier otro medicamento IBP o a cualquiera de los ingredientes de PROTONIX. Consulte el final de esta Guía del medicamento para obtener una lista completa de ingredientes.
- tomando un medicamento que contiene rilpivirina (EDURANT, COMPLERA, ODEFSEY, JULUCA) utilizado para tratar el VIH-1 (virus de inmunodeficiencia humana).
Antes de tomar PROTONIX, informe a su médico acerca de todas sus condiciones médicas, incluso si usted:
- tiene niveles bajos de magnesio en la sangre.
- está embarazada o planea quedar embarazada. PROTONIX 40 mg puede dañar a su bebé por nacer. Informe a su médico si se queda embarazada o cree que podría estarlo durante el tratamiento con PROTONIX.
- están amamantando o planean amamantar. PROTONIX puede pasar a la leche materna. Hable con su médico sobre la mejor manera de alimentar a su bebé si toma PROTONIX.
Informe a su médico sobre todos los medicamentos que toma, incluyendo medicamentos recetados y de venta libre, vitaminas y suplementos a base de hierbas. Informe especialmente a su médico si toma metotrexato (Otrexup, Rasuvo, Trexall, XATMEP), digoxina (LANOXIN) o un diurético.
¿Cómo debo tomar PROTONIX 20mg?
- Tome PROTONIX 40 mg exactamente según lo prescrito por su médico.
PROTONIX comprimidos de liberación retardada (PROTONIX comprimidos de 20 mg):
-
- No parta, mastique ni triture las tabletas de PROTONIX de 40 mg.
- Trague los comprimidos de PROTONIX 20 mg enteros, con o sin alimentos.
- Informe a su médico si no puede tragar su comprimido de 20 mg de PROTONIX.
- Puede usar antiácidos mientras toma tabletas de 40 mg de PROTONIX.
PROTONIX 40 mg para suspensión oral de liberación retardada (PROTONIX para suspensión oral):
-
- No parta, mastique ni triture PROTONIX 20 mg para suspensión oral.
- Tome PROTONIX 40 mg para suspensión oral unos 30 minutos antes de una comida.
- PROTONIX para suspensión oral solo debe administrarse por vía oral mezclado con jugo de manzana o puré de manzana o a través de una sonda nasogástrica (NG) o una sonda de gastrostomía mezclado con jugo de manzana. No mezcle PROTONIX para suspensión oral en líquidos que no sean jugo de manzana o alimentos que no sean compota de manzana.
- No divida un paquete de PROTONIX 20 mg para suspensión oral para hacer una dosis más pequeña.
- Ver el "Instrucciones de uso" al final de esta Guía del medicamento para obtener instrucciones sobre cómo mezclar y tomar PROTONIX 40 mg para suspensión oral por vía oral en puré de manzana o jugo de manzana o cómo mezclar y administrar la suspensión a través de una sonda nasogástrica o una sonda de gastrostomía mezclada con jugo de manzana.
- Si olvida una dosis de PROTONIX, tómela lo antes posible. Si es casi la hora de su próxima dosis, no tome la dosis olvidada. Tome la siguiente dosis a la hora habitual. No tome 2 dosis al mismo tiempo.
- Si toma demasiado PROTONIX, llame a su médico o al centro de toxicología al 1-800-222-1222 de inmediato o diríjase a la sala de emergencias más cercana.
¿Cuáles son los posibles efectos secundarios de PROTONIX 40mg?
PROTONIX 20 mg puede causar efectos secundarios graves, que incluyen:
- Ver “¿Cuál es la información más importante que debo saber sobre PROTONIX?”
- Niveles bajos de vitamina B-12 en su cuerpo puede ocurrir en personas que han tomado PROTONIX 40 mg durante mucho tiempo (más de 3 años). Informe a su médico si tiene síntomas de niveles bajos de vitamina B-12, que incluyen dificultad para respirar, mareos, latidos cardíacos irregulares, debilidad muscular, piel pálida, sensación de cansancio, cambios de humor y hormigueo o entumecimiento en los brazos y las piernas.
- Niveles bajos de magnesio en su cuerpo puede ocurrir en personas que han tomado PROTONIX durante al menos 3 meses. Informe a su médico si tiene síntomas de niveles bajos de magnesio, como convulsiones, mareos, latidos cardíacos irregulares, nerviosismo, dolores musculares o debilidad y espasmos en las manos, los pies o la voz.
- Crecimientos estomacales (pólipos de glándulas fúndicas). Las personas que toman medicamentos PPI durante mucho tiempo tienen un mayor riesgo de desarrollar cierto tipo de crecimientos estomacales llamados pólipos de las glándulas fúndicas, especialmente después de tomar medicamentos PPI durante más de 1 año.
Los efectos secundarios más comunes de PROTONIX 40 mg en adultos incluyen: dolor de cabeza, diarrea, náuseas, dolor en el área del estómago (abdominal), vómitos, gases, mareos y dolor en las articulaciones.
Los efectos secundarios más comunes de PROTONIX 40 mg en niños incluyen: infección de las vías respiratorias superiores, dolor de cabeza, fiebre, diarrea, vómitos, sarpullido y dolor en el área del estómago (abdominal). Estos no son todos los posibles efectos secundarios de PROTONIX. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo conservar PROTONIX 40mg?
Guarde PROTONIX 20 mg a temperatura ambiente, entre 20 °C y 25 °C (68 °F y 77 °F).
Mantenga PROTONIX y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de PROTONIX.
A veces, los medicamentos se recetan para fines distintos a los enumerados en la Guía del medicamento. No use PROTONIX para una condición para la cual no fue recetado. No le dé PROTONIX 20 mg a otras personas, incluso si tienen los mismos síntomas que usted. Puede que les haga daño. Puede pedirle a su médico o farmacéutico información sobre PROTONIX 20 mg que está escrita para profesionales de la salud.
¿Cuáles son los ingredientes de PROTONIX?
Ingrediente activo: pantoprazol sódico sesquihidrato
Ingredientes inactivos en las tabletas de liberación retardada de PROTONIX: estearato de calcio, crospovidona, hipromelosa, óxido de hierro, manitol, copolímero de ácido metacrílico, polisorbato 80, povidona, propilenglicol, carbonato de sodio, lauril sulfato de sodio, dióxido de titanio y citrato de trietilo.
Ingredientes inactivos en PROTONIX 40 mg para suspensión oral de liberación retardada: crospovidona, hipromelosa, copolímero de ácido metacrílico, celulosa microcristalina, polisorbato 80, povidona, carbonato de sodio, lauril sulfato de sodio, talco, dióxido de titanio, citrato de trietilo y óxido férrico amarillo.
Instrucciones de uso
PROTONIX (pro-TAH-nix) (pantoprazol sódico) para suspensión oral de liberación retardada
PROTONIX 40 mg de liberación retardada suspensión oral (PROTONIX 40 mg para suspensión oral):
Información importante:
- No parta, mastique ni triture PROTONIX para suspensión oral.
- Tome PROTONIX para suspensión oral unos 30 minutos antes de una comida.
- PROTONIX para suspensión oral:
- debería solo debe tomarse con puré de manzana o jugo de manzana.
- debería no se mezcle con agua u otros líquidos, ni con otros alimentos.
- El paquete no debe dividirse para hacer una dosis más pequeña.
Tomar PROTONIX 20 mg para suspensión oral con compota de manzana:
Tomar PROTONIX 40 mg para suspensión oral con jugo de manzana:
Administración de PROTONIX 40 mg para suspensión oral a través de una sonda nasogástrica (NG) o una sonda de gastrostomía:
- PROTONIX para suspensión oral se puede administrar a través de una sonda NG o una sonda de gastrostomía que se tamaño 16 francés o más grande. No administre PROTONIX 20 mg para suspensión oral a través de una sonda NG o una sonda de gastrostomía de tamaño inferior a 16 French.
- Mezcla PROTONIX 20 mg para suspensión oral solo en jugo de manzana cuando se administra a través de una sonda NG o una sonda de gastrostomía.
¿Cómo debo almacenar PROTONIX 20mg?
Guarde PROTONIX 20 mg a temperatura ambiente, entre 20 °C y 25 °C (68 °F y 77 °F).
Mantenga PROTONIX 40 mg y todos los medicamentos fuera del alcance de los niños.
Esta Guía del medicamento y las Instrucciones de uso han sido aprobadas por la Administración de Alimentos y Medicamentos de EE. UU.