Uniphyl 400mg Theophylline Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Uniphyl 400 mg y cómo se usa?
Uniphyl es un medicamento recetado que se usa para tratar los síntomas del asma, la bronquitis, el enfisema y otros problemas respiratorios (broncoespasmo agudo). Uniphyl 400 mg se puede usar solo o con otros medicamentos.
Uniphyl pertenece a una clase de medicamentos llamados derivados de xantina; Inhibidores de la enzima fosfodiesterasa, no selectivos.
No se sabe si Uniphyl 400 mg es seguro y efectivo en niños menores de 1 ½ meses de edad.
¿Cuáles son los posibles efectos secundarios de Uniphyl?
Uniphyl puede causar efectos secundarios graves que incluyen:
- urticaria,
- respiración dificultosa,
- hinchazón de la cara, los labios, la lengua o la garganta,
- vómitos severos o continuos,
- dolor de cabeza continuo,
- problemas para dormir,
- latidos rápidos del corazón,
- embargo,
- fiebre,
- calambres en las piernas,
- estreñimiento,
- latidos irregulares del corazón,
- revoloteando en tu pecho,
- aumento de la sed o la micción,
- entumecimiento u hormigueo,
- debilidad muscular,
- sensación de cojera,
- aumento de la sed,
- aumento de la micción,
- boca seca, y
- aliento con olor afrutado
Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Uniphyl incluyen:
- náuseas,
- vómitos,
- Diarrea,
- dolor de cabeza,
- problemas para dormir (insomnio),
- temblores,
- transpiración,
- inquietud, y
- irritabilidad
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Uniphyl. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
DESCRIPCIÓN
Uniphyl (tableta de teofilina anhidra) ® (teofilina, anhidra) Las tabletas en un sistema de liberación controlada permiten un intervalo de dosificación de 24 horas para los pacientes apropiados.
La teofilina se clasifica estructuralmente como una metilxantina. Se presenta como un polvo blanco, inodoro y cristalino con un sabor amargo.
La teofilina anhidra tiene el nombre químico 1H-Purine-2,6-dione, 3,7-dihydro-1,3-dimethyl-, y está representada por la siguiente fórmula estructural:
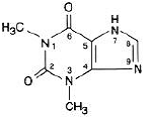
La fórmula molecular de la teofilina anhidra es C7H8N4O2 con un peso molecular de 180,17.
Cada tableta de liberación controlada para administración oral contiene 400 o 600 mg de teofilina anhidra.
Ingredientes inactivos: alcohol cetoestearílico, hidroxietilcelulosa, estearato de magnesio, povidona y talco.
INDICACIONES
La teofilina está indicada para el tratamiento de los síntomas y la obstrucción reversible del flujo de aire asociados con el asma crónica y otras enfermedades pulmonares crónicas, por ejemplo, enfisema y bronquitis crónica.
DOSIFICACIÓN Y ADMINISTRACIÓN
Uniphyl (tableta de teofilina anhidra) ® 400 o 600 mg Las tabletas se pueden tomar una vez al día por la mañana o por la noche. Se recomienda que Uniphyl (tableta de teofilina anhidra) se tome con las comidas. Se debe advertir a los pacientes que si eligen tomar Uniphyl (tableta anhidra de teofilina) con alimentos, deben tomarlo de manera constante con alimentos y si lo toman en ayunas, deben tomarlo en ayunas de manera rutinaria. Es importante que el producto siempre que se dosifique se dosifique consistentemente con o sin alimentos.
Uniphyl (tableta de teofilina anhidra) ® Las tabletas no deben masticarse ni triturarse porque puede provocar una liberación rápida de teofilina con potencial de toxicidad. La tableta ranurada se puede partir. Con poca frecuencia, los pacientes que reciben Uniphyl (tabletas de teofilina anhidra) de 400 o 600 mg pueden expulsar una tableta de matriz intacta en las heces o por colostomía. Estas tabletas de matriz generalmente contienen poca o ninguna teofilina residual.
Los pacientes estabilizados, de 12 años de edad o mayores, que estén tomando un producto de teofilina de liberación inmediata o de liberación controlada pueden ser transferidos a la administración una vez al día de 400 mg o 600 mg de Uniphyl (tableta de teofilina anhidra) Tabletas en una dosis de mg por mg base.
Debe reconocerse que los niveles máximos y mínimos de teofilina sérica producidos por la dosificación una vez al día pueden variar de los producidos por el producto y/o régimen anterior.
Consideraciones Generales
La concentración sérica máxima de teofilina en estado estacionario es una función de la dosis, el intervalo de dosificación y la tasa de absorción y eliminación de teofilina en el paciente individual. Debido a las marcadas diferencias individuales en la tasa de depuración de teofilina, la dosis requerida para lograr una concentración sérica máxima de teofilina en el rango de 10-20 mcg/mL varía cuatro veces entre pacientes por lo demás similares en ausencia de factores que se sabe que alteran la depuración de teofilina (p. 400-1600 mg/día en adultos La dosis de teofilina debe individualizarse sobre la base de las mediciones de la concentración sérica máxima de teofilina para lograr una dosis que brinde el máximo beneficio potencial con el mínimo riesgo de efectos adversos.
Los efectos adversos transitorios similares a la cafeína y las concentraciones séricas excesivas en los metabolizadores lentos se pueden evitar en la mayoría de los pacientes comenzando con una dosis lo suficientemente baja y aumentando lentamente la dosis, si se considera clínicamente indicado, en pequeños incrementos (Ver ). Los aumentos de dosis solo se deben realizar si la dosis anterior es bien tolerada y en intervalos de no menos de 3 días para permitir que las concentraciones séricas de teofilina alcancen el nuevo estado estacionario. El ajuste de la dosis debe guiarse por la medición de la concentración de teofilina sérica (ver PRECAUCIONES , Pruebas de laboratorio y DOSIFICACIÓN Y ADMINISTRACIÓN ). Los proveedores de atención médica deben instruir a los pacientes y cuidadores para que descontinúen cualquier dosis que cause efectos adversos, suspendan el medicamento hasta que estos síntomas desaparezcan y luego reanude la terapia a una dosis más baja tolerada previamente (ver ADVERTENCIAS ).
Si los síntomas del paciente están bien controlados, no hay efectos adversos aparentes ni factores intervinientes que puedan alterar los requisitos de dosificación (ver ADVERTENCIAS y PRECAUCIONES ), las concentraciones de teofilina sérica deben controlarse a intervalos de 6 meses para los niños que crecen rápidamente y a intervalos anuales para todos los demás. En pacientes gravemente enfermos, las concentraciones séricas de teofilina deben controlarse a intervalos frecuentes, por ejemplo, cada 24 horas.
La teofilina se distribuye mal en la grasa corporal, por lo tanto, la dosis en mg/kg debe calcularse sobre la base del peso corporal ideal.
La Tabla V contiene el esquema de titulación de dosis de teofilina recomendado para pacientes en varios grupos de edad y circunstancias clínicas.
La Tabla VI contiene recomendaciones para el ajuste de la dosis de teofilina en función de las concentraciones séricas de teofilina. La aplicación de estas recomendaciones posológicas generales a pacientes individuales debe tener en cuenta las características clínicas únicas de cada paciente. En general, estas recomendaciones deberían servir como el límite superior para los ajustes de dosis a fin de disminuir el riesgo de eventos adversos potencialmente graves asociados con grandes aumentos inesperados en la concentración de teofilina sérica.
B. Pacientes con factores de riesgo para el aclaramiento alterado, ancianos (> 60 años) y aquellos en quienes no es factible monitorear las concentraciones séricas de teofilina:
En niños de 12 a 15 años, la dosis de teofilina no debe exceder los 16 mg/kg/día hasta un máximo de 400 mg/día en presencia de factores de riesgo de eliminación reducida de teofilina (ver ADVERTENCIAS ) o si no es factible monitorear las concentraciones de teofilina sérica.
En adolescentes ≥ 16 años y adultos, incluidos los ancianos, la dosis de teofilina no debe exceder los 400 mg/día en presencia de factores de riesgo de eliminación reducida de teofilina (ver ADVERTENCIAS ) o si no es factible monitorear las concentraciones de teofilina sérica.
*Los pacientes con un metabolismo más rápido identificados clínicamente por requisitos de dosis superiores a la media deben recibir una dosis más pequeña con mayor frecuencia (cada 12 horas) para evitar síntomas de avance resultantes de concentraciones mínimas bajas antes de la siguiente dosis.CUADRO VI. Ajuste de dosis guiado por la concentración de teofilina sérica.
CÓMO SUMINISTRADO
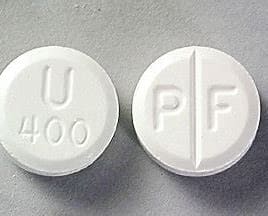
Uniphyl® (teofilina, anhidra) tabletas de liberación controlada 400 mg se presentan en frascos de plástico blanco, opaco, a prueba de niños, que contienen 100 comprimidos ( CDN 67781-251-01) o 500 tabletas ( CDN 67781-251-05). Cada comprimido blanco redondo de 400 mg lleva el símbolo PF en el lado ranurado y U400 en el otro lado.
Uniphyl® (teofilina, anhidra) tabletas de liberación controlada 600 mg se presentan en frascos de plástico blanco, opaco, a prueba de niños, que contienen 100 comprimidos ( CDN 67781-252-01). Cada comprimido rectangular, cóncavo, blanco de 600 mg lleva el símbolo PF en el lado ranurado y U 600 en el otro lado.
Almacenar a 25°C (77°F); excursiones permitidas entre 15°-30°C (59°-86°F).
Dispensar en un recipiente hermético resistente a la luz.
Productos farmacéuticos Purdue LP, Dist. por: Purdue Pharmaceutical Products LP, Stamford, CT 06901-3431. 17 de marzo de 2004.EFECTOS SECUNDARIOS
Las reacciones adversas asociadas con la teofilina son generalmente leves cuando las concentraciones séricas máximas de teofilina son SOBREDOSIS ). Las reacciones adversas transitorias similares a la cafeína ocurren en aproximadamente el 50 % de los pacientes cuando la terapia con teofilina se inicia en dosis más altas que las dosis iniciales recomendadas (p. ej., > 300 mg/día en adultos y > 12 mg/kg/día en niños mayores de > 1 año de edad). ). Durante el inicio de la terapia con teofilina, los efectos adversos similares a la cafeína pueden alterar transitoriamente el comportamiento del paciente, especialmente en niños en edad escolar, pero esta respuesta rara vez persiste. El inicio de la terapia con teofilina a una dosis baja con posterior titulación lenta hasta una dosis máxima predeterminada relacionada con la edad reducirá significativamente la frecuencia de estos efectos adversos transitorios (ver DOSIFICACIÓN Y ADMINISTRACIÓN ). En un pequeño porcentaje de pacientes (
Otras reacciones adversas que se han informado con concentraciones séricas de teofilina
CUADRO IV. Manifestaciones de toxicidad por teofilina. *
INTERACCIONES CON LA DROGAS
La teofilina interactúa con una amplia variedad de fármacos. La interacción puede ser farmacodinámica, es decir, alteraciones en la respuesta terapéutica a la teofilina u otro fármaco o aparición de efectos adversos sin un cambio en la concentración de teofilina sérica. Sin embargo, con más frecuencia, la interacción es farmacocinética, es decir, la velocidad de eliminación de la teofilina se ve alterada por otro fármaco, lo que da como resultado un aumento o disminución de las concentraciones séricas de teofilina. La teofilina rara vez altera la farmacocinética de otros fármacos. Los medicamentos enumerados en la Tabla II tienen el potencial de producir interacciones farmacodinámicas o farmacocinéticas clínicamente significativas con la teofilina. La información en la columna "Efecto" de la Tabla II asume que el fármaco que interactúa se agrega a un régimen de teofilina en estado estacionario. Si se inicia el tratamiento con teofilina en un paciente que ya está tomando un fármaco que inhibe la depuración de teofilina (p. ej., cimetidina, eritromicina), la dosis de teofilina necesaria para lograr una concentración sérica terapéutica de teofilina será menor. Por el contrario, si se inicia el tratamiento con teofilina en un paciente que ya está tomando un fármaco que aumenta la depuración de teofilina (p. ej., rifampicina), la dosis de teofilina requerida para lograr una concentración sérica terapéutica de teofilina será mayor. La suspensión de un fármaco concomitante que aumente la depuración de teofilina dará como resultado una acumulación de teofilina a niveles potencialmente tóxicos, a menos que la dosis de teofilina se reduzca adecuadamente. La suspensión de un fármaco concomitante que inhiba la depuración de teofilina dará como resultado una disminución de las concentraciones séricas de teofilina, a menos que la dosis de teofilina se incremente adecuadamente. Se ha documentado que los medicamentos enumerados en la Tabla III no interactúan con la teofilina o no producen una interacción clínicamente significativa (es decir,
La lista de medicamentos en las Tablas II y III está actualizada al 9 de febrero de 1995. Continuamente se informan nuevas interacciones para la teofilina, especialmente con nuevas entidades químicas. El profesional de la salud no debe asumir que un fármaco no interactúa con la teofilina si no figura en la Tabla II. Antes de agregar un fármaco recientemente disponible en un paciente que recibe teofilina, se debe consultar el prospecto del nuevo fármaco y/o la literatura médica para determinar si se ha informado una interacción entre el nuevo fármaco y la teofilina.
TABLA II. Interacciones farmacológicas clínicamente significativas con teofilina.*
CUADRO III. Medicamentos que se ha documentado que no interactúan con la teofilina o medicamentos que no producen una interacción clínicamente significativa con la teofilina. *
Interacciones entre medicamentos y alimentos
Se ha estudiado la biodisponibilidad de Uniphyl® Tablets (teofilina, anhidra) con la administración concomitante de alimentos. En tres estudios de dosis única, los sujetos que recibieron Uniphyl (comprimidos de teofilina anhidra) de 400 mg o 600 mg con una comida estandarizada rica en grasas se compararon con condiciones en ayunas. En condiciones de alimentación, la concentración plasmática máxima y la biodisponibilidad aumentaron; sin embargo, no fue evidente un aumento precipitado en la tasa y el grado de absorción (ver Farmacocinética, Absorción ). El aumento del pico y la extensión de la absorción en condiciones de alimentación sugiere que la dosificación debe administrarse idealmente de forma consistente con o sin alimentos.
El efecto de otras drogas en las mediciones de concentración sérica de teofilina
La mayoría de los ensayos de teofilina sérica en uso clínico son inmunoensayos que son específicos para la teofilina. Estos ensayos no detectan otras xantinas como la cafeína, la difilina y la pentoxifilina. Sin embargo, algunos fármacos (p. ej., cefazolina, cefalotina) pueden interferir con ciertas técnicas de HPLC. Los metabolitos de cafeína y xantina en recién nacidos o pacientes con disfunción renal pueden hacer que la lectura de algunos métodos de oficina con reactivos secos sea más alta que la concentración real de teofilina sérica.
ADVERTENCIAS
Enfermedad concurrente
La teofilina debe usarse con extrema precaución en pacientes con las siguientes condiciones clínicas debido al mayor riesgo de exacerbación de la condición concurrente:
Enfermedad ulcerosa péptica activa Trastornos convulsivos Arritmias cardíacas (sin incluir bradiarritmias)
Condiciones que reducen la eliminación de teofilina
Hay varias causas fácilmente identificables de eliminación reducida de teofilina. Si la dosis diaria total no se reduce adecuadamente en presencia de estos factores de riesgo, puede ocurrir una toxicidad grave y potencialmente mortal por teofilina. Se debe considerar cuidadosamente los beneficios y riesgos del uso de teofilina y la necesidad de un control más intensivo de las concentraciones séricas de teofilina en pacientes con los siguientes factores de riesgo:
Años
Recién nacidos (a término y prematuros) Niños 60 años)
Enfermedades concurrentes
Edema pulmonar agudo Insuficiencia cardiaca congestiva Cor-pulmonale Fiebre; ≥ 102° durante 24 horas o más; o menores elevaciones de temperatura por períodos más largos Hipotiroidismo Enfermedad del hígado; cirrosis, hepatitis aguda Disminución de la función renal en lactantes
dejar de fumar
Interacciones con la drogas
Añadir un fármaco que inhiba el metabolismo de la teofilina (p. ej., cimetidina, eritromicina, tacrina) o suspender un fármaco administrado simultáneamente que aumente el metabolismo de la teofilina (p. ej., carbamazepina, rifampicina). (Ver PRECAUCIONES: INTERACCIONES MEDICAMENTOSAS , ).
Cuando hay signos o síntomas de toxicidad por teofilina
Cada vez que un paciente que recibe teofilina desarrolle náuseas o vómitos, particularmente vómitos repetitivos, u otros signos o síntomas consistentes con la toxicidad de la teofilina (incluso si se sospecha otra causa), se deben suspender las dosis adicionales de teofilina y se debe medir la concentración sérica de teofilina de inmediato. Se debe indicar a los pacientes que no continúen con ninguna dosis que cause efectos adversos y que suspendan las dosis posteriores hasta que los síntomas hayan desaparecido, momento en el cual el profesional de la salud puede indicarle al paciente que reanude el medicamento a una dosis más baja (ver DOSIFICACIÓN Y ADMINISTRACIÓN , Pautas de dosificación, Tabla VI ).
Aumentos de dosis
No se deben realizar aumentos en la dosis de teofilina en respuesta a una exacerbación aguda de los síntomas de la enfermedad pulmonar crónica, ya que la teofilina brinda pocos beneficios adicionales a los agonistas selectivos beta2 inhalados y los corticosteroides administrados sistémicamente en esta circunstancia y aumenta el riesgo de efectos adversos. Debe medirse una concentración sérica máxima de teofilina en estado estacionario antes de aumentar la dosis en respuesta a síntomas crónicos persistentes para determinar si es seguro aumentar la dosis. Antes de aumentar la dosis de teofilina en base a una concentración sérica baja, el profesional sanitario debe considerar si la muestra de sangre se obtuvo en el momento adecuado en relación con la dosis y si el paciente se ha adherido al régimen prescrito (ver PRECAUCIONES , Pruebas de laboratorio ).
Dado que la tasa de eliminación de teofilina puede depender de la dosis (es decir, las concentraciones séricas en estado estacionario pueden aumentar de manera desproporcionada al aumento de la dosis), un aumento de la dosis basado en una medición de la concentración sérica subterapéutica debe ser conservador. En general, limitar los aumentos de dosis a alrededor del 25 % de la dosis diaria total anterior reducirá el riesgo de aumentos excesivos no intencionados en la concentración de teofilina sérica (ver DOSIFICACIÓN Y ADMINISTRACIÓN , ).
PRECAUCIONES
General
Antes de iniciar el tratamiento con teofilina, antes de aumentar la dosis de teofilina y durante el seguimiento (ver ADVERTENCIAS ). La dosis de teofilina seleccionada para el inicio de la terapia debe ser baja y, si se tolera, debe aumentarse lentamente durante un período de una semana o más con la dosis final guiada por el control de las concentraciones séricas de teofilina y la respuesta clínica del paciente (ver DOSIFICACIÓN Y ADMINISTRACIÓN , ).
Monitoreo de las concentraciones de teofilina sérica
Las mediciones de la concentración de teofilina sérica están fácilmente disponibles y deben usarse para determinar si la dosis es adecuada. Específicamente, la concentración de teofilina sérica debe medirse de la siguiente manera:
Para guiar un aumento de la dosis, la muestra de sangre debe obtenerse en el momento de la concentración máxima esperada de teofilina en suero; 12 horas después de una dosis vespertina o 9 horas después de una dosis matutina en estado estacionario. Para la mayoría de los pacientes, el estado de equilibrio se alcanzará después de 3 días de dosificación cuando no se hayan omitido dosis, no se hayan agregado dosis adicionales y ninguna de las dosis se haya tomado a intervalos desiguales. Una concentración mínima (es decir, al final del intervalo de dosificación) no proporciona información útil adicional y puede dar lugar a un aumento inadecuado de la dosis, ya que la concentración sérica máxima de teofilina puede ser dos o más veces mayor que la concentración mínima con una formulación de liberación inmediata. . Si la muestra de suero se extrae más de 12 horas después de la dosis de la noche, o más de 9 horas después de la dosis de la mañana, los resultados deben interpretarse con precaución ya que la concentración puede no reflejar la concentración máxima. Por el contrario, cuando hay signos o síntomas de toxicidad por teofilina, se debe obtener una muestra de suero lo antes posible, analizarla de inmediato y comunicar el resultado al profesional sanitario sin demora. En pacientes en los que se sospeche una disminución de la unión a proteínas séricas (p. ej., cirrosis, mujeres durante el tercer trimestre del embarazo), se debe medir la concentración de teofilina libre y ajustar la dosis para lograr una concentración libre de 6-12 mcg/mL. Las concentraciones de saliva de teofilina no se pueden usar de manera confiable para ajustar la dosis sin técnicas especiales.
Efectos en las pruebas de laboratorio
Como resultado de sus efectos farmacológicos, la teofilina en concentraciones séricas dentro del rango de 10-20 mcg/mL aumenta modestamente la glucosa plasmática (de una media de 88 mg% a 98 mg%), el ácido úrico (de una media de 4 mg/dL a 6 mg/dL), ácidos grasos libres (de una media de 451 μEq/L a 800 μEq/L), colesterol total (de una media de 140 vs 160 mg/dL), HDL (de una media de 36 a 50 mg /dL), relación HDL/LDL (de una media de 0,5 a 0,7) y excreción urinaria de cortisol libre (de una media de 44 a 63 mcg/24 h) Teofilina en concentraciones séricas dentro del rango de 10-20 mcg/mL también puede disminuir transitoriamente las concentraciones séricas de triyodotironina (144 antes, 131 después de una semana y 142 ng/dL después de 4 semanas de teofilina).La importancia clínica de estos cambios debe sopesarse frente al beneficio terapéutico potencial de la teofilina en pacientes individuales.
Carcinogénesis, mutagénesis y deterioro de la fertilidad
Se han realizado estudios de carcinogenicidad a largo plazo en ratones (dosis orales de 30 a 150 mg/kg) y ratas (dosis orales de 5 a 75 mg/kg). Los resultados están pendientes.
La teofilina se ha estudiado en Ames salmonella, citogenética in vivo e in vitro, micronúcleos y sistemas de prueba de ovario de hámster chino y no se ha demostrado que sea genotóxica.
En un estudio de reproducción continuo de 14 semanas, la teofilina, administrada a parejas de ratones B6C3F1 en apareamiento en dosis orales de 120, 270 y 500 mg/kg (aproximadamente 1,0-3,0 veces la dosis humana en mg/m²), perjudicó la fertilidad, como lo demuestra la disminuciones en el número de crías vivas por camada, disminuciones en el número medio de camadas por pareja fértil y aumentos en el período de gestación con la dosis alta, así como disminuciones en la proporción de crías nacidas vivas con la dosis media y alta. En estudios de toxicidad de 13 semanas, se administró teofilina a ratas F344 y ratones B6C3F1 en dosis orales de 40-300 mg/kg (aproximadamente 2,0 veces la dosis humana en mg/m²). A la dosis alta, se observó toxicidad sistémica en ambas especies, incluida la disminución del peso testicular.
El embarazo
Efectos teratogénicos: Categoría C
En estudios en los que se administraron dosis a ratones, ratas y conejas preñadas durante el período de organogénesis, la teofilina produjo efectos teratogénicos.
En estudios con ratones, una dosis intraperitoneal única de 100 mg/kg o más (aproximadamente igual a la dosis oral máxima recomendada para adultos en mg/m²) durante la organogénesis produjo paladar hendido y anomalías digitales. Se observaron micromelia, micrognatia, pie zambo, hematoma subcutáneo, párpados abiertos y embrioletalidad a dosis que son aproximadamente 2 veces la dosis oral máxima recomendada para adultos en mg/m².
En un estudio con ratas que recibieron dosis desde la concepción hasta la organogénesis, una dosis oral de 150 mg/kg/día (aproximadamente 2 veces la dosis oral máxima recomendada para adultos en mg/m²) produjo anomalías digitales. Se observó embrioletalidad con una dosis subcutánea de 200 mg/kg/día (aproximadamente 4 veces la dosis oral máxima recomendada para adultos sobre una base de mg/m²). En un estudio en el que se dosificó a conejas preñadas durante la organogénesis, una dosis intravenosa de 60 mg/kg/día (aproximadamente 2 veces la dosis oral máxima recomendada para adultos en mg/m²), que causó la muerte de una hembra y síntomas clínicos. signos en otros, produjo paladar hendido y fue embrioletal. Las dosis de 15 mg/kg/día y superiores (menos de la dosis oral máxima recomendada para adultos en mg/m²) aumentaron la incidencia de variaciones esqueléticas.
No existen estudios adecuados y bien controlados en mujeres embarazadas. La teofilina debe usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Madres lactantes
La teofilina se excreta en la leche materna y puede causar irritabilidad u otros signos de toxicidad leve en lactantes humanos. La concentración de teofilina en la leche materna es aproximadamente equivalente a la concentración sérica materna. Un bebé que ingiere un litro de leche materna que contiene 10-20 mcg/mL de teofilina por día probablemente reciba 10-20 mg de teofilina por día. Es improbable que se produzcan efectos adversos graves en el lactante, a menos que la madre tenga concentraciones séricas tóxicas de teofilina.
Uso pediátrico
La teofilina es segura y eficaz para las indicaciones aprobadas en pacientes pediátricos. La dosis de mantenimiento de teofilina debe seleccionarse con precaución en pacientes pediátricos, ya que la tasa de eliminación de teofilina es muy variable en el rango de edad pediátrica (ver FARMACOLOGÍA CLÍNICA , , ADVERTENCIAS , y DOSIFICACIÓN Y ADMINISTRACIÓN , ).
Uso geriátrico
Los pacientes de edad avanzada tienen un riesgo significativamente mayor de experimentar toxicidad grave por la teofilina que los pacientes más jóvenes debido a los cambios farmacocinéticos y farmacodinámicos asociados con el envejecimiento. El aclaramiento de teofilina se reduce en un promedio del 30 % en adultos mayores sanos (> 60 años) en comparación con adultos jóvenes sanos. El aclaramiento de teofilina puede reducirse aún más por enfermedades concomitantes predominantes en los ancianos, que deterioran aún más el aclaramiento de este fármaco y tienen el potencial de aumentar los niveles séricos y la toxicidad potencial. Estas condiciones incluyen deterioro de la función renal, enfermedad pulmonar obstructiva crónica, insuficiencia cardíaca congestiva, enfermedad hepática y una mayor prevalencia del uso de ciertos medicamentos (ver PRECAUCIONES : INTERACCIONES CON LA DROGAS ) con el potencial de interacción farmacocinética y farmacodinámica. La unión a proteínas puede disminuir en los ancianos, lo que resulta en una mayor proporción de la concentración sérica total de teofilina en la forma no unida farmacológicamente activa. Los pacientes de edad avanzada también parecen ser más sensibles a los efectos tóxicos de la teofilina después de una sobredosis crónica que los pacientes más jóvenes. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes de edad avanzada (ver PRECAUCIONES , Monitoreo de las concentraciones de teofilina sérica , y DOSIFICACIÓN Y ADMINISTRACIÓN ).
La dosis diaria máxima de teofilina en pacientes mayores de 60 años normalmente no debe exceder los 400 mg/día, a menos que el paciente continúe sintomático y la concentración sérica máxima de teofilina en el estado estacionario sea DOSIFICACIÓN Y ADMINISTRACIÓN ). Las dosis de teofilina superiores a 400 mg/d deben prescribirse con precaución en pacientes de edad avanzada.
SOBREDOSIS
General
La cronicidad y el patrón de sobredosis de teofilina influyen significativamente en las manifestaciones clínicas de toxicidad, manejo y resultado. Hay dos presentaciones comunes: (1) sobredosis aguda, es decir, la ingestión de una sola dosis grande excesiva (> 10 mg/kg), como ocurre en el contexto de un intento de suicidio o error de medicación aislado, y (2) sobredosis crónica, es decir, la ingestión de dosis repetidas que son excesivas para la tasa de eliminación de teofilina del paciente. Las causas más comunes de sobredosis crónica de teofilina incluyen el error del paciente o del cuidador en la dosificación, la prescripción por parte del profesional de la salud de una dosis excesiva o una dosis normal en presencia de factores que se sabe que reducen la tasa de eliminación de teofilina y el aumento de la dosis en respuesta a una exacerbación. de los síntomas sin medir primero la concentración de teofilina sérica para determinar si es seguro aumentar la dosis.
La toxicidad severa por sobredosis de teofilina es un evento relativamente raro. En una organización de mantenimiento de la salud, la frecuencia de ingresos hospitalarios por sobredosis crónica de teofilina fue de aproximadamente 1 por 1000 años-persona de exposición. En otro estudio, entre 6000 muestras de sangre obtenidas para la medición de la concentración de teofilina sérica, por cualquier motivo, de pacientes tratados en un departamento de emergencias, el 7 % estaba en el rango de 20-30 mcg/mL y el 3 % era > 30 mcg/mL. Aproximadamente dos tercios de los pacientes con concentraciones séricas de teofilina en el rango de 20-30 mcg/mL tuvieron una o más manifestaciones de toxicidad mientras que > 90% de los pacientes con concentraciones séricas de teofilina > 30 mcg/mL estaban clínicamente intoxicados. De manera similar, en otros informes, la toxicidad grave de la teofilina se observa principalmente en concentraciones séricas > 30 mcg/mL.
Varios estudios han descrito las manifestaciones clínicas de la sobredosis de teofilina y han intentado determinar los factores que predicen una toxicidad potencialmente mortal. En general, los pacientes que experimentan una sobredosis aguda tienen menos probabilidades de sufrir convulsiones que los pacientes que han sufrido una sobredosis crónica, a menos que la concentración sérica máxima de teofilina sea > 100 mcg/ml. Después de una sobredosis crónica, pueden ocurrir convulsiones generalizadas, arritmias cardíacas potencialmente mortales y la muerte a concentraciones séricas de teofilina > 30 mcg/mL. La gravedad de la toxicidad después de una sobredosis crónica se correlaciona más fuertemente con la edad del paciente que con la concentración sérica máxima de teofilina; los pacientes > 60 años corren el mayor riesgo de toxicidad grave y mortalidad después de una sobredosis crónica. La enfermedad preexistente o concurrente también puede aumentar significativamente la susceptibilidad de un paciente a una manifestación tóxica particular, por ejemplo, los pacientes con trastornos neurológicos tienen un mayor riesgo de convulsiones y los pacientes con enfermedad cardíaca tienen un mayor riesgo de arritmias cardíacas para una concentración de teofilina sérica determinada en comparación con a pacientes sin la enfermedad de base.
La frecuencia de diversas manifestaciones notificadas de sobredosis de teofilina según el modo de sobredosis se enumeran en la Tabla IV. Otras manifestaciones de la toxicidad de la teofilina incluyen aumentos en el calcio sérico, creatina quinasa, mioglobina y recuento de leucocitos, disminución de fosfato y magnesio séricos, infarto agudo de miocardio y retención urinaria en hombres con uropatía obstructiva. Las convulsiones asociadas con concentraciones séricas de teofilina > 30 mcg/mL a menudo son resistentes a la terapia anticonvulsiva y pueden provocar lesiones cerebrales irreversibles si no se controlan rápidamente. La muerte por toxicidad por teofilina es más a menudo secundaria a un paro cardiorrespiratorio y/o encefalopatía hipóxica después de convulsiones generalizadas prolongadas o arritmias cardíacas intratables que causan compromiso hemodinámico.
Manejo de sobredosis
Recomendaciones generales para pacientes con síntomas de sobredosis de teofilina o concentraciones séricas de teofilina > 30 mcg/mL (Nota: las concentraciones séricas de teofilina pueden continuar aumentando después de la presentación del paciente para atención médica).
Al mismo tiempo que instaura el tratamiento, comuníquese con un centro regional de toxicología para obtener información actualizada y asesoramiento sobre cómo individualizar las recomendaciones que siguen.
Instituir atención de apoyo, incluido el establecimiento de un acceso intravenoso, el mantenimiento de las vías respiratorias y la monitorización electrocardiográfica.
Tratamiento de las convulsiones
Debido a la alta morbilidad y mortalidad asociadas con las convulsiones inducidas por teofilina, el tratamiento debe ser rápido y agresivo. La terapia anticonvulsiva debe iniciarse con una benzodiazepina intravenosa, por ejemplo, diazepam, en incrementos de 0,1 a 0,2 mg/kg cada 1 a 3 minutos hasta que se detengan las convulsiones. Las convulsiones repetitivas deben tratarse con una dosis de carga de fenobarbital (20 mg/kg infundidos durante 30 a 60 minutos). Los informes de casos de sobredosis de teofilina en humanos y estudios en animales sugieren que la fenitoína no es efectiva para terminar con las convulsiones inducidas por teofilina. Las dosis de benzodiazepinas y fenobarbital requeridas para terminar con las convulsiones inducidas por teofilina están cerca de las dosis que pueden causar depresión respiratoria grave o paro respiratorio; por lo tanto, el profesional de la salud debe estar preparado para proporcionar ventilación asistida. Los pacientes de edad avanzada y los pacientes con EPOC pueden ser más susceptibles a los efectos depresores respiratorios de los anticonvulsivos. Puede ser necesario el coma inducido por barbitúricos o la administración de anestesia general para terminar con las convulsiones repetitivas o el estado epiléptico. La anestesia general debe usarse con precaución en pacientes con sobredosis de teofilina porque los anestésicos volátiles fluorados pueden sensibilizar el miocardio a las catecolaminas endógenas liberadas por la teofilina. El enflurano parece menos probable que esté asociado con este efecto que el halotano y, por lo tanto, puede ser más seguro. Los bloqueadores neuromusculares solos no deben usarse para terminar las convulsiones ya que suprimen las manifestaciones musculoesqueléticas sin terminar la actividad convulsiva en el cerebro.
Anticipe la necesidad de anticonvulsivos
En pacientes con sobredosis de teofilina que tienen un alto riesgo de convulsiones inducidas por teofilina, por ejemplo, pacientes con sobredosis aguda y concentraciones séricas de teofilina > 100 mcg/mL o sobredosis crónica en pacientes > 60 años de edad con concentraciones séricas de teofilina > 30 mcg/mL , se debe anticipar la necesidad de terapia anticonvulsiva. Una benzodiazepina como el diazepam debe introducirse en una jeringa y mantenerse al lado de la cama del paciente y el personal médico calificado para tratar las convulsiones debe estar disponible de inmediato. En pacientes seleccionados con alto riesgo de convulsiones inducidas por teofilina, se debe considerar la administración de una terapia anticonvulsiva profiláctica. Las situaciones en las que se debe considerar la terapia anticonvulsiva profiláctica en pacientes de alto riesgo incluyen demoras anticipadas en el establecimiento de métodos para la extracción extracorpórea de teofilina (p. ej., traslado de un paciente de alto riesgo de un centro de atención médica a otro para la extracción extracorpórea) y circunstancias clínicas que interfieren significativamente con los esfuerzos para aumentar la depuración de teofilina (p. ej., un recién nacido en el que la diálisis puede no ser técnicamente factible o un paciente con vómitos que no responde a los antieméticos que no puede tolerar múltiples dosis de carbón activado por vía oral). En estudios con animales, se ha demostrado que la administración profiláctica de fenobarbital, pero no de fenitoína, retrasa la aparición de convulsiones generalizadas inducidas por teofilina y aumenta la dosis de teofilina requerida para inducir convulsiones (es decir, aumenta notablemente la LD50). Aunque no existen estudios controlados en humanos, una dosis de carga de fenobarbital intravenoso (20 mg/kg infundidos durante 60 minutos) puede retrasar o prevenir convulsiones potencialmente mortales en pacientes de alto riesgo mientras continúan los esfuerzos para mejorar la depuración de teofilina. El fenobarbital puede causar depresión respiratoria, particularmente en pacientes de edad avanzada y pacientes con EPOC.
Tratamiento de las arritmias cardiacas
La taquicardia sinusal y los extrasístoles ventriculares simples no son precursores de arritmias potencialmente mortales, no requieren tratamiento en ausencia de compromiso hemodinámico y se resuelven con la disminución de las concentraciones séricas de teofilina. Otras arritmias, especialmente aquellas asociadas con compromiso hemodinámico, deben tratarse con terapia antiarrítmica apropiada para el tipo de arritmia.
Descontaminación gastrointestinal
El carbón activado por vía oral (0,5 g/kg hasta 20 g y repetir al menos una vez 1-2 horas después de la primera dosis) es extremadamente eficaz para bloquear la absorción de teofilina en el tracto gastrointestinal, incluso cuando se administra varias horas después de la ingestión. Si el paciente está vomitando, el carbón debe administrarse a través de una sonda nasogástrica o después de la administración de un antiemético. Deben evitarse los antieméticos de fenotiazina, como la proclorperazina o la perfenazina, ya que pueden reducir el umbral convulsivo y, con frecuencia, causar reacciones distónicas. Se puede usar una dosis única de sorbitol para promover la defecación y facilitar la eliminación de la teofilina unida al carbón del tracto gastrointestinal. Sin embargo, el sorbitol debe dosificarse con precaución ya que es un potente purgante que puede causar alteraciones profundas de líquidos y electrolitos, particularmente después de múltiples dosis. Las combinaciones fijas comercialmente disponibles de carbón líquido y sorbitol deben evitarse en niños pequeños y después de la primera dosis en adolescentes y adultos, ya que no permiten la individualización de la dosificación de carbón y sorbitol. El jarabe de ipecacuana debe evitarse en sobredosis de teofilina. Aunque la ipecacuana induce emesis, no reduce la absorción de teofilina a menos que se administre dentro de los 5 minutos posteriores a la ingestión e incluso entonces es menos eficaz que el carbón activado por vía oral. Además, la emesis inducida por la ipecacuana puede persistir durante varias horas después de una sola dosis y disminuir significativamente la retención y la eficacia del carbón activado por vía oral.
Teofilina sérica
Monitoreo de la concentración La concentración de teofilina sérica debe medirse inmediatamente después de la presentación, 2 a 4 horas después y luego a intervalos suficientes, por ejemplo, cada 4 horas, para guiar las decisiones de tratamiento y evaluar la efectividad de la terapia. Las concentraciones séricas de teofilina pueden continuar aumentando después de la presentación del paciente para atención médica como resultado de la absorción continua de teofilina en el tracto gastrointestinal. El monitoreo en serie de las concentraciones séricas de teofilina en suero debe continuar hasta que quede claro que la concentración ya no aumenta y ha regresado a niveles no tóxicos.
Monitoreo general
Procedimientos La monitorización electrocardiográfica debe iniciarse en el momento de la presentación y continuarse hasta que el nivel de teofilina sérica haya vuelto a un nivel no tóxico. Los electrolitos séricos y la glucosa deben medirse en el momento de la presentación y en los intervalos apropiados indicados por las circunstancias clínicas. Las anormalidades de líquidos y electrolitos deben corregirse rápidamente. El seguimiento y el tratamiento deben continuarse hasta que la concentración sérica disminuya por debajo de 20 mcg/mL.
Mejorar la eliminación de la teofilina
El carbón activado oral en dosis múltiples (p. ej., 0,5 mg/kg hasta 20 g, cada dos horas) aumenta el aclaramiento de teofilina al menos dos veces por adsorción de la teofilina secretada en los fluidos gastrointestinales. El carbón debe retenerse y pasar a través del tracto gastrointestinal para que sea efectivo; Por lo tanto, la emesis debe controlarse mediante la administración de antieméticos apropiados. Alternativamente, el carbón vegetal se puede administrar de forma continua a través de una sonda nasogástrica junto con los antieméticos apropiados. Se puede administrar una sola dosis de sorbitol con el carbón activado para promover la defecación y facilitar la eliminación de la teofilina adsorbida del tracto gastrointestinal. El sorbitol por sí solo no mejora la eliminación de la teofilina y debe dosificarse con precaución para evitar la deposición excesiva que puede provocar desequilibrios graves de líquidos y electrolitos. Las combinaciones fijas comercialmente disponibles de carbón líquido y sorbitol deben evitarse en niños pequeños y después de la primera dosis en adolescentes y adultos, ya que no permiten la individualización de la dosificación de carbón y sorbitol. En pacientes con vómitos intratables, se deben instituir métodos extracorpóreos de eliminación de teofilina (ver SOBREDOSIS , Eliminación extracorpórea ).
Recomendaciones específicas
Sobredosis aguda
Sobredosis crónica
Eliminación extracorpórea
El aumento de la tasa de eliminación de teofilina por métodos extracorpóreos puede disminuir rápidamente las concentraciones séricas, pero los riesgos del procedimiento deben sopesarse frente al beneficio potencial. La hemoperfusión con carbón vegetal es el método más efectivo de extracción extracorpórea, aumentando el aclaramiento de teofilina hasta seis veces, pero pueden ocurrir complicaciones graves, como hipotensión, hipocalcemia, consumo de plaquetas y diátesis hemorrágica. La hemodiálisis es casi tan eficiente como el carbón activado oral en dosis múltiples y tiene un menor riesgo de complicaciones graves que la hemoperfusión con carbón. La hemodiálisis se debe considerar como una alternativa cuando la hemoperfusión con carbón no es factible y las dosis múltiples de carbón oral son ineficaces debido a la emesis intratable. Las concentraciones séricas de teofilina pueden recuperarse de 5 a 10 mcg/ml después de interrumpir la hemoperfusión con carbón o la hemodiálisis debido a la redistribución de la teofilina desde el compartimento tisular. La diálisis peritoneal es ineficaz para eliminar la teofilina; las exanguinotransfusiones en recién nacidos han sido mínimamente efectivas.
CONTRAINDICACIONES
Uniphyl (comprimido anhidro de teofilina) ® está contraindicado en pacientes con antecedentes de hipersensibilidad a la teofilina u otros componentes del producto.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
La teofilina tiene dos acciones distintas en las vías respiratorias de pacientes con obstrucción reversible; relajación del músculo liso (es decir, broncodilatación) y supresión de la respuesta de las vías respiratorias a los estímulos (es decir, efectos profilácticos no broncodilatadores). Si bien los mecanismos de acción de la teofilina no se conocen con certeza, los estudios en animales sugieren que la broncodilatación está mediada por la inhibición de dos isoenzimas de la fosfodiesterasa (PDE III y, en menor medida, PDE IV), mientras que las acciones profilácticas no broncodilatadoras son probablemente mediada por uno o más mecanismos moleculares diferentes, que no implican la inhibición de la PDE III o el antagonismo de los receptores de adenosina. Algunos de los efectos adversos asociados con la teofilina parecen estar mediados por la inhibición de la PDE III (p. ej., hipotensión, taquicardia, dolor de cabeza y emesis) y el antagonismo del receptor de adenosina (p. ej., alteraciones en el flujo sanguíneo cerebral).
La teofilina aumenta la fuerza de contracción de los músculos diafragmáticos. Esta acción parece deberse a la mejora de la captación de calcio a través de un canal mediado por adenosina.
Relación concentración sérica-efecto
La broncodilatación ocurre en el rango de concentración de teofilina sérica de 5-20 mcg/mL. En la mayoría de los estudios se ha encontrado una mejora clínicamente importante en el control de los síntomas que requiere concentraciones séricas máximas de teofilina > 10 mcg/mL, pero los pacientes con enfermedad leve pueden beneficiarse de concentraciones más bajas. A concentraciones séricas de teofilina > 20 mcg/ml, aumentan tanto la frecuencia como la gravedad de las reacciones adversas. En general, mantener las concentraciones séricas máximas de teofilina entre 10 y 15 mcg/mL logrará la mayor parte del beneficio terapéutico potencial del fármaco y minimizará el riesgo de eventos adversos graves.
Farmacocinética
Visión general
La teofilina se absorbe rápida y completamente después de la administración oral en solución o en forma de dosificación oral sólida de liberación inmediata. La teofilina no sufre ninguna eliminación presistémica apreciable, se distribuye libremente en los tejidos libres de grasa y se metaboliza ampliamente en el hígado.
La farmacocinética de la teofilina varía ampliamente entre pacientes similares y no puede predecirse por edad, sexo, peso corporal u otras características demográficas. Además, ciertas enfermedades concurrentes y alteraciones en la fisiología normal (ver Tabla I) y la coadministración de otras drogas (ver Tabla II) pueden alterar significativamente las características farmacocinéticas de la teofilina. La variabilidad dentro del sujeto en el metabolismo también se ha informado en algunos estudios, especialmente en pacientes con enfermedades agudas. Por lo tanto, se recomienda medir las concentraciones séricas de teofilina con frecuencia en pacientes gravemente enfermos (p. ej., a intervalos de 24 horas) y periódicamente en pacientes que reciben terapia a largo plazo, p. ej., a intervalos de 6 a 12 meses. Se deben realizar mediciones más frecuentes en presencia de cualquier condición que pueda alterar significativamente el aclaramiento de teofilina (ver PRECAUCIONES, Pruebas de Laboratorio ).
TABLA I. Media y rango del aclaramiento corporal total y la vida media de la teofilina en relación con la edad y los estados fisiológicos alterados.¶
Nota: Además de los factores enumerados anteriormente, la eliminación de teofilina aumenta y la vida media disminuye con dietas bajas en carbohidratos/altas en proteínas, nutrición parenteral y consumo diario de carne de res asada al carbón. Una dieta alta en carbohidratos y baja en proteínas puede disminuir la eliminación y prolongar la vida media de la teofilina.
Absorción
Uniphyl (comprimido anhidro de teofilina) ® administrado con alimentos se absorbe completamente después de la administración oral.
En un estudio cruzado de dosis única, se administraron dos tabletas de Uniphyl (teofilina anhidra) de 400 mg a 19 voluntarios normales por la mañana o por la noche inmediatamente después de la misma comida estandarizada (769 calorías que consisten en 97 gramos de carbohidratos, 33 gramos de proteínas y 27 gramos de grasa). No hubo evidencia de dumping de dosis ni hubo diferencias significativas en los parámetros farmacocinéticos atribuibles al momento de la administración del fármaco. En el brazo de la mañana, los parámetros farmacocinéticos fueron AUC = 241,9 ± 83,0 mcg hr/mL, Cmax = 9,3 ± 2,0 mcg/mL, Tmax = 12,8 ± 4,2 horas. En el brazo de la noche, los parámetros farmacocinéticos fueron AUC = 219,7 ± 83,0 mcg h/mL, Cmax = 9,2 ± 2,0 mcg/mL, Tmax = 12,5 ± 4,2 horas.
Un estudio en el que se administraron tabletas de 400 mg de Uniphyl (teofilina anhidra) a 17 asmáticos adultos alimentados produjo curvas de tiempo-nivel de teofilina similares cuando se administró por la mañana o por la noche. Los niveles séricos fueron generalmente más altos en el régimen vespertino, pero no hubo diferencias estadísticamente significativas entre los dos regímenes.
Un estudio de dosis única en 15 voluntarios masculinos normales en ayunas cuya vida media de eliminación media inherente de teofilina se verificó mediante un producto de teofilina líquida en 6,9 ± 2,5 (SD) horas se les administró dos o tres tabletas de 400 mg de Uniphyl (tableta anhidra de teofilina) ® . La biodisponibilidad relativa de Uniphyl (comprimido anhidro de teofilina) administrado en ayunas en comparación con un producto de liberación inmediata fue del 59 %. Los niveles máximos de teofilina en suero se produjeron a las 6,9 ± 5,2 (SD) horas, con un nivel máximo normalizado (a 800 mg) de 6,2 ± 2,1 (SD). La vida media de eliminación aparente para las tabletas de 400 mg de Uniphyl (tableta anhidra de teofilina) fue de 17,2 ± 5,8 (DE) horas.
La farmacocinética en estado estacionario se determinó en un estudio en 12 pacientes en ayunas con enfermedad pulmonar obstructiva crónica reversible. Todos recibieron dos tabletas de Uniphyl (teofilina anhidra) de 400 mg administradas una vez al día por la mañana y un producto BID de liberación controlada de referencia administrado como dos tabletas de 200 mg administradas con 12 horas de diferencia. Los parámetros farmacocinéticos obtenidos para los comprimidos de Uniphyl (teofilina anhidra) administrados en dosis de 800 mg una vez al día por la mañana fueron prácticamente idénticos a los parámetros correspondientes para el fármaco de referencia cuando se administró en dosis de 400 mg dos veces al día. En particular, los valores de AUC, Cmax y Cmin obtenidos en este estudio fueron los siguientes:
Los estudios de dosis única en los que los sujetos estuvieron en ayunas durante doce (12) horas antes y cuatro (4) horas adicionales después de la dosificación, demostraron una biodisponibilidad reducida en comparación con la dosificación con alimentos. Un estudio de dosis única en 20 voluntarios normales que recibieron dos (2) tabletas de 400 mg por la mañana, comparó la dosis en estas condiciones de ayuno con la dosis inmediatamente antes de un desayuno estandarizado (769 calorías, que consisten en 97 gramos de carbohidratos, 33 gramos de proteína y 27 gramos de grasa). En condiciones de alimentación, los parámetros farmacocinéticos fueron: AUC = 231,7 ± 92,4 mcg hr/mL, Cmax = 8,4 ± 2,6 mcg/mL, Tmax = 17,3 ± 6,7 horas. En condiciones de ayuno, estos parámetros fueron AUC = 141,2 ± 6,53 mcg hr/mL, Cmax = 5,5 ± 1,5 mcg/mL, Tmax = 6,5 ± 2,1 horas.
Otro estudio de dosis única en 21 voluntarios masculinos normales, dosificados por la noche, comparó el ayuno con una comida estandarizada alta en calorías y grasas (870-1,020 calorías, que consta de 33 gramos de proteína, 55-75 gramos de grasa, 58 gramos de carbohidratos). En el brazo de ayuno, los sujetos recibieron una tableta de Uniphyl (teofilina anhidra) ® de 400 mg a las 8 pm después de un ayuno de ocho horas seguido de otro ayuno de cuatro horas. En el brazo alimentado, los sujetos recibieron nuevamente una dosis de 400 mg de Uniphyl (tableta de teofilina anhidra), pero a las 8 pm inmediatamente después de la comida estandarizada con alto contenido de grasa citada anteriormente. Los parámetros farmacocinéticos (normalizados a 800 mg) administrados fueron AUC = 221,8 ± 40,9 mcg hr/mL, Cmax = 10,9 ± 1,7 mcg/mL, Tmax = 11,8 ± 2,2 horas. En el brazo en ayunas, los parámetros farmacocinéticos (normalizados a 800 mg) fueron AUC = 146,4 ± 40,9 mcg hr/mL, Cmax = 6,7 ± 1,7 mcg/mL, Tmax = 7,3 ± 2,2 horas.
Por lo tanto, la administración de dosis únicas de Uniphyl (tableta de teofilina anhidra) a voluntarios normales sanos, en ayunas prolongadas (al menos 10 horas de ayuno nocturno antes de la dosificación seguido de un ayuno adicional de cuatro (4) horas después de la dosificación) da como resultado una disminución de la biodisponibilidad. Sin embargo, no hubo fallas en este sistema de administración que condujeran a una liberación repentina e inesperada de una gran cantidad de teofilina con Uniphyl (tabletas de teofilina anhidra), incluso cuando se administraron con una comida rica en grasas y calorías.
Se realizaron estudios similares con la tableta de 600 mg de Uniphyl (tableta de teofilina anhidra). Un estudio de dosis única en 24 sujetos con un aclaramiento de teofilina establecido de ≤ 4 L/h, comparó la evaluación farmacocinética de una tableta de 600 mg de Uniphyl (tableta de teofilina anhidra) y una tableta y media de 400 mg de Uniphyl (tableta de teofilina anhidra) bajo alimentación (usando una dieta alta en grasas estándar) y condiciones de ayuno. Los resultados de este estudio cruzado aleatorizado de 4 vías demuestran la bioequivalencia de las tabletas de Uniphyl (tableta de teofilina anhidra) de 400 mg y 600 mg. En condiciones de alimentación, los resultados farmacocinéticos para una tableta y media de 400 mg fueron AUC = 214,64 ± 55,88 mcg hr/mL, Cmax = 10,58 ± 2,21 mcg/mL y Tmax = 9,00 ± 2,64 horas, y para la tableta de 600 mg fueron AUC = 207,85 ± 48,9 mcg hr/mL, Cmax = 10,39 ± 1,91 mcg/mL y Tmax = 9,58 ± 1,86 horas. En condiciones de ayuno, los resultados farmacocinéticos para una tableta y media de 400 mg fueron AUC = 191,85 ± 51,1 mcg hr/mL, Cmax = 7,37 ± 1,83 mcg/mL y Tmax = 8,08 ± 4,39 horas; y para la tableta de 600 mg fueron AUC = 199,39 ± 70,27 mcg hr/mL, Cmax = 7,66 ± 2,09 mcg/mL y Tmax = 9,67 ± 4,89 horas.
En este estudio, las proporciones promedio de alimentación/ayuno para una tableta y media de 400 mg y la tableta de 600 mg fueron de aproximadamente 112 % y 104 %, respectivamente.
En otro estudio, se examinó la biodisponibilidad de la tableta de Uniphyl (tableta de teofilina anhidra) de 600 mg con la administración por la mañana y por la noche. Este estudio cruzado de dosis única en 22 hombres sanos se realizó en condiciones de alimentación (dieta estándar alta en grasas). Los resultados no demostraron ninguna diferencia clínicamente significativa en la biodisponibilidad de la tableta de 600 mg de Uniphyl (tableta de teofilina anhidra) administrada por la mañana o por la noche. Los resultados fueron: AUC = 233,6 ± 45,1 mcg hr/mL, Cmax = 10,6 ± 1,3 mcg/mL y Tmax = 12,5 ± 3,2 horas con dosis matutina; AUC = 209,8 ± 46,2 mcg hr/mL, Cmax = 9,7 ± 1,4 mcg/mL y Tmax = 13,7 ± 3,3 horas con dosis vespertinas. La relación PM/AM fue del 89,3%.
Las características de absorción de las tabletas Uniphyl® (teofilina, anhidra) se han estudiado ampliamente. Un estudio cruzado de biodisponibilidad en estado estacionario en 22 hombres normales comparó dos tabletas de Uniphyl (teofilina anhidra) de 400 mg administradas cada 24 horas a las 8 am inmediatamente después del desayuno con un producto de teofilina de liberación controlada de referencia administrado dos veces al día en sujetos alimentados a las 8 am inmediatamente después del desayuno y 8 pm inmediatamente después de la cena (769 calorías, que consisten en 97 gramos de carbohidratos, 33 gramos de proteína y 27 gramos de grasa).
Los parámetros farmacocinéticos de Uniphyl (tableta de teofilina anhidra) de 400 mg en estas condiciones de estado estacionario fueron AUC = 203,3 ± 87,1 mcg h/mL, Cmax = 12,1 ± 3,8 mcg/mL, Cmin = 4,50 ± 3,6, Tmax = 8,8 ± 4,6 horas. Para el producto BID de referencia, los parámetros farmacocinéticos fueron AUC = 219,2 ± 88,4 mcg hr/mL, Cmax = 11,0 ± 4,1 mcg/mL, Cmin = 7,28 ± 3,5, Tmax = 6,9 ± 3,4 horas. La fluctuación porcentual media [(Cmax-Cmin/Cmin)x100] = 169 % para el régimen de una vez al día y 51 % para el régimen BID del producto de referencia.
La biodisponibilidad de la tableta de 600 mg de Uniphyl (tableta de teofilina anhidra) se evaluó más a fondo en un estudio de estado estacionario de dosis múltiples en 26 hombres sanos que comparó la tableta de 600 mg con una tableta y media de 400 mg de Uniphyl (tableta de teofilina anhidra). Todos los sujetos habían establecido previamente aclaramientos de teofilina de ≤ 4 l/h y se les administró una dosis diaria durante 6 días en condiciones de alimentación. Los resultados no mostraron diferencias clínicamente significativas entre los regímenes de tabletas de 600 mg y uno y medio de 400 mg de Uniphyl (tableta de teofilina anhidra). Los resultados del estado estacionario fueron:
La relación de biodisponibilidad para los comprimidos de 600/400 mg fue del 98,8 %. Por lo tanto, bajo todas las condiciones del estudio, la tableta de 600 mg es bioequivalente a una tableta y media de 400 mg.
Los estudios demuestran que siempre que los sujetos se alimentaran o ayunaran constantemente, existe una biodisponibilidad similar con la administración de Uniphyl (tableta de teofilina anhidra) una vez al día, ya sea por la mañana o por la noche.
Distribución
Una vez que la teofilina ingresa a la circulación sistémica, alrededor del 40% se une a las proteínas plasmáticas, principalmente a la albúmina. La teofilina libre se distribuye por todo el agua corporal, pero se distribuye pobremente en la grasa corporal. El volumen aparente de distribución de la teofilina es de aproximadamente 0,45 L/kg (rango 0,3-0,7 L/kg) basado en el peso corporal ideal. La teofilina pasa libremente a través de la placenta, a la leche materna y al líquido cefalorraquídeo (LCR). Las concentraciones de teofilina en saliva se aproximan a las concentraciones séricas no unidas, pero no son fiables para el control terapéutico o de rutina a menos que se utilicen técnicas especiales. Se produce un aumento en el volumen de distribución de la teofilina, principalmente debido a la reducción de la unión a proteínas plasmáticas, en recién nacidos prematuros, pacientes con cirrosis hepática, acidemia no corregida, ancianos y mujeres durante el tercer trimestre del embarazo. En tales casos, el paciente puede mostrar signos de toxicidad a concentraciones séricas totales (ligadas + no ligadas) de teofilina en el rango terapéutico (10-20 mcg/mL) debido a concentraciones elevadas del fármaco no ligado farmacológicamente activo. De manera similar, un paciente con unión de teofilina disminuida puede tener una concentración total de fármaco subterapéutica mientras que la concentración no unida farmacológicamente activa está en el rango terapéutico. Si solo se mide la concentración sérica total de teofilina, esto puede dar lugar a un aumento de la dosis innecesario y potencialmente peligroso. En pacientes con unión reducida a proteínas, la medición de la concentración de teofilina sérica libre proporciona un medio más confiable de ajuste de dosis que la medición de la concentración total de teofilina sérica. En general, las concentraciones de teofilina libre deben mantenerse en el rango de 6 a 12 mcg/mL.
Metabolismo
Después de la dosificación oral, la teofilina no sufre ninguna eliminación de primer paso medible. En adultos y niños mayores de un año, aproximadamente el 90% de la dosis se metaboliza en el hígado. La biotransformación tiene lugar mediante la desmetilación a 1-metilxantina y 3-metilxantina y la hidroxilación a ácido 1,3-dimetilúrico. La 1-metilxantina se hidroxila aún más, mediante la xantina oxidasa, a ácido 1-metilúrico. Alrededor del 6% de una dosis de teofilina está N-metilada a cafeína. La desmetilación de la teofilina a 3-metilxantina es catalizada por el citocromo P-450 1A2, mientras que los citocromos P-450 2E1 y P-450 3A3 catalizan la hidroxilación a ácido 1,3-dimetilúrico. La desmetilación a 1-metilxantina parece ser catalizada por el citocromo P-450 1A2 o por un citocromo estrechamente relacionado. En los recién nacidos, la vía de N-desmetilación está ausente mientras que la función de la vía de hidroxilación es marcadamente deficiente. La actividad de estas vías aumenta lentamente hasta niveles máximos al año de edad.
La cafeína y la 3-metilxantina son los únicos metabolitos de teofilina con actividad farmacológica. La 3-metilxantina tiene aproximadamente una décima parte de la actividad farmacológica de la teofilina y las concentraciones séricas en adultos con función renal normal son
Tanto la vía de N-desmetilación como la de hidroxilación de la biotransformación de la teofilina tienen una capacidad limitada. Debido a la amplia variabilidad interindividual de la tasa de metabolismo de la teofilina, la eliminación no lineal puede comenzar en algunos pacientes con concentraciones séricas de teofilina DOSIS Y ADMINISTRACION, Cuadro VI ). No es posible predecir con precisión la dependencia de la dosis del metabolismo de la teofilina en pacientes a priori, pero los pacientes con índices de depuración iniciales muy altos (es decir, concentraciones bajas de teofilina sérica en estado estacionario a dosis superiores al promedio) tienen la mayor probabilidad de experimentar grandes cambios en los niveles séricos. concentración de teofilina en respuesta a los cambios de dosis.
Excreción
En los recién nacidos, aproximadamente el 50 % de la dosis de teofilina se excreta sin cambios en la orina. Más allá de los primeros tres meses de vida, aproximadamente el 10% de la dosis de teofilina se excreta sin cambios en la orina. El resto se excreta en la orina principalmente como ácido 1,3-dimetilúrico (35-40 %), ácido 1-metilúrico (20-25 %) y 3-metilxantina (15-20 %). Debido a que poca teofilina se excreta sin cambios en la orina y dado que los metabolitos activos de la teofilina (es decir, cafeína, 3-metilxantina) no se acumulan a niveles clínicamente significativos incluso en caso de enfermedad renal en etapa terminal, no es necesario ajustar la dosis para la insuficiencia renal. en adultos y niños > 3 meses de edad. Por el contrario, la gran fracción de la dosis de teofilina excretada en la orina como teofilina sin cambios y cafeína en los recién nacidos requiere una cuidadosa atención a la reducción de la dosis y un control frecuente de las concentraciones séricas de teofilina en los recién nacidos con función renal reducida (Ver ADVERTENCIAS ).
Concentraciones séricas en estado estacionario
Después de múltiples dosis de teofilina, el estado estacionario se alcanza en 30 a 65 horas (promedio de 40 horas) en adultos. En estado estacionario, con un régimen de dosificación con intervalos de 24 horas, la concentración mínima media esperada es aproximadamente el 50 % de la concentración máxima media, suponiendo una vida media media de teofilina de 8 horas. La diferencia entre las concentraciones máxima y mínima es mayor en pacientes con un aclaramiento de teofilina más rápido. En estos pacientes la administración de Uniphyl (tableta de teofilina anhidra) ® puede requerirse con mayor frecuencia (cada 12 horas).
Poblaciones especiales (Consulte la Tabla I para conocer los valores medios de depuración y vida media)
geriátrico
El aclaramiento de teofilina se reduce en un promedio del 30 % en adultos mayores sanos (> 60 años) en comparación con adultos jóvenes sanos. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes de edad avanzada (ver ADVERTENCIAS ).
Pediatría
El aclaramiento de la teofilina es muy bajo en los recién nacidos (ver ADVERTENCIAS ). El aclaramiento de teofilina alcanza valores máximos al año de edad, permanece relativamente constante hasta alrededor de los 9 años y luego disminuye lentamente en aproximadamente un 50 % a los valores de adultos alrededor de los 16 años. La excreción renal de teofilina sin cambios en los recién nacidos asciende a alrededor del 50 % dosis, en comparación con alrededor del 10% en niños mayores de tres meses y en adultos. En los pacientes pediátricos se requiere una cuidadosa atención a la selección de la dosis y el control de las concentraciones séricas de teofilina (ver ADVERTENCIAS y DOSIFICACIÓN Y ADMINISTRACIÓN ).
Género
Las diferencias de género en el aclaramiento de teofilina son relativamente pequeñas y es poco probable que tengan importancia clínica. Sin embargo, se ha informado una reducción significativa en la depuración de teofilina en mujeres en el día 20 del ciclo menstrual y durante el tercer trimestre del embarazo.
La raza
No se han estudiado las diferencias farmacocinéticas en el aclaramiento de teofilina debido a la raza.
Insuficiencia renal
Sólo una pequeña fracción, por ejemplo, alrededor del 10%, de la dosis de teofilina administrada se excreta sin cambios en la orina de niños mayores de tres meses y adultos. Debido a que poca teofilina se excreta sin cambios en la orina y dado que los metabolitos activos de la teofilina (es decir, cafeína, 3-metilxantina) no se acumulan a niveles clínicamente significativos incluso en caso de enfermedad renal en etapa terminal, no es necesario ajustar la dosis para la insuficiencia renal en adultos. y niños > 3 meses de edad. Por el contrario, aproximadamente el 50% de la dosis de teofilina administrada se excreta sin cambios en la orina de los recién nacidos. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en recién nacidos con función renal disminuida (ver ADVERTENCIAS ).
insuficiencia hepática
El aclaramiento de teofilina se reduce en un 50% o más en pacientes con insuficiencia hepática (p. ej., cirrosis, hepatitis aguda, colestasis). Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes con función hepática reducida (ver ADVERTENCIAS ).
Insuficiencia cardíaca congestiva (CHF)
El aclaramiento de teofilina se reduce en un 50% o más en pacientes con ICC. El grado de reducción del aclaramiento de teofilina en pacientes con ICC parece estar directamente relacionado con la gravedad de la enfermedad cardiaca. Dado que la depuración de teofilina es independiente del flujo sanguíneo hepático, la reducción de la depuración parece deberse a una función alterada de los hepatocitos más que a una perfusión reducida. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes con ICC (ver ADVERTENCIAS ).
fumadores
Fumar tabaco y marihuana parece aumentar la depuración de teofilina por inducción de vías metabólicas. Se ha demostrado que el aclaramiento de teofilina aumenta en aproximadamente un 50 % en fumadores de tabaco adultos jóvenes y en aproximadamente un 80 % en fumadores de tabaco de edad avanzada en comparación con sujetos no fumadores. También se ha demostrado que la exposición pasiva al humo aumenta la eliminación de teofilina hasta en un 50 %. La abstinencia de fumar tabaco durante una semana provoca una reducción de aproximadamente un 40% en el aclaramiento de teofilina. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes que dejan de fumar (ver ADVERTENCIAS ). Se ha demostrado que el uso de chicles de nicotina no tiene efecto sobre la depuración de teofilina.
Fiebre
La fiebre, independientemente de su causa subyacente, puede disminuir la eliminación de teofilina. La magnitud y la duración de la fiebre parecen estar directamente relacionadas con el grado de disminución del aclaramiento de teofilina. Se carece de datos precisos, pero probablemente se requiera una temperatura de 39 °C (102 °F) durante al menos 24 horas para producir un aumento clínicamente significativo en las concentraciones de teofilina sérica. Los niños con tasas rápidas de depuración de teofilina (es decir, aquellos que requieren una dosis sustancialmente mayor que el promedio [p. ej., > 22 mg/kg/día] para lograr una concentración máxima terapéutica de teofilina sérica cuando no tienen fiebre) pueden tener un mayor riesgo de efectos tóxicos. efectos de la disminución de la depuración durante la fiebre sostenida. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones séricas de teofilina en pacientes con fiebre sostenida (ver ADVERTENCIAS ).
Misceláneas
Otros factores asociados con la disminución del aclaramiento de teofilina incluyen el tercer trimestre del embarazo, sepsis con falla multiorgánica e hipotiroidismo. Se requiere atención cuidadosa a la reducción de la dosis y monitoreo frecuente de las concentraciones de teofilina sérica en pacientes con cualquiera de estas condiciones (ver ADVERTENCIAS ). Otros factores asociados con el aumento de la depuración de teofilina incluyen el hipertiroidismo y la fibrosis quística.
Estudios clínicos
En pacientes con asma crónica, incluidos pacientes con asma grave que requieren corticosteroides inhalados o corticosteroides orales en días alternos, muchos estudios clínicos han demostrado que la teofilina disminuye la frecuencia y la gravedad de los síntomas, incluidas las exacerbaciones nocturnas, y disminuye el uso "según sea necesario" de corticosteroides inhalados. agonistas beta-2. También se ha demostrado que la teofilina reduce la necesidad de cursos cortos de prednisona oral diaria para aliviar las exacerbaciones de la obstrucción de las vías respiratorias que no responden a los broncodilatadores en los asmáticos.
En pacientes con enfermedad pulmonar obstructiva crónica (EPOC), los estudios clínicos han demostrado que la teofilina disminuye la disnea, el atrapamiento de aire, el trabajo respiratorio y mejora la contractilidad de los músculos diafragmáticos con poca o ninguna mejora en las mediciones de la función pulmonar.
INFORMACIÓN DEL PACIENTE
Se debe indicar al paciente (o padre/cuidador) que consulte a un médico siempre que presente náuseas, vómitos, dolor de cabeza persistente, insomnio o latidos cardíacos rápidos durante el tratamiento con teofilina, incluso si se sospecha otra causa. Se debe instruir al paciente para que se comunique con su profesional de la salud si desarrolla una nueva enfermedad, especialmente si se acompaña de fiebre persistente, si experimenta un empeoramiento de una enfermedad crónica, si comienza o deja de fumar cigarrillos o marihuana, o si otro profesional de la salud agrega un nuevo medicamento o descontinúa un medicamento previamente recetado. Se debe informar a los pacientes que la teofilina interactúa con una amplia variedad de fármacos (ver Tabla II). El suplemento dietético Hierba de San Juan (Hypericum perforatum) no debe tomarse al mismo tiempo que la teofilina, ya que puede provocar una disminución de los niveles de teofilina. Si los pacientes ya están tomando St. John's Wort y teofilina juntos, deben consultar a su profesional de la salud antes de dejar de tomar St. John's Wort, ya que sus concentraciones de teofilina pueden aumentar cuando se hace esto, lo que resulta en toxicidad. Se debe instruir a los pacientes para que informen a todos los profesionales de la salud involucrados en su atención que están tomando teofilina, especialmente cuando se agrega o elimina un medicamento de su tratamiento. Se debe instruir a los pacientes para que no modifiquen la dosis, el momento de la dosis o la frecuencia de administración sin consultar primero a su profesional de la salud. Si se olvida una dosis, se debe indicar al paciente que tome la siguiente dosis a la hora programada habitualmente y que no intente compensar la dosis olvidada.
Las tabletas Uniphyl (teofilina anhidra) ® se pueden tomar una vez al día por la mañana o por la noche. Se recomienda que Uniphyl (tableta de teofilina anhidra) se tome con las comidas. Se debe advertir a los pacientes que si eligen tomar Uniphyl (tableta anhidra de teofilina) con alimentos, deben tomarlo de manera constante con alimentos y si lo toman en ayunas, deben tomarlo en ayunas de manera rutinaria. Es importante que el producto siempre que se dosifique se dosifique consistentemente con o sin alimentos.
Uniphyl (tableta anhidra de teofilina) Las tabletas no deben masticarse ni triturarse porque puede provocar una liberación rápida de teofilina con potencial de toxicidad. La tableta ranurada se puede partir. Los pacientes que reciben tabletas de Uniphyl (teofilina anhidra) pueden expulsar una tableta de matriz intacta en las heces o por colostomía. Estas tabletas de matriz generalmente contienen poca o ninguna teofilina residual.