Rebetol 200mg Ribavirin Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Rebetol 200mg y cómo se usa?
Rebetol es un medicamento recetado que se usa para tratar los síntomas de la hepatitis C crónica. Rebetol 200 mg se puede usar solo o con otros medicamentos.
Rebetol pertenece a una clase de medicamentos llamados Agentes de Hepatitis B/Hepatitis C; Agentes RSV.
No se sabe si Rebetol 200 mg es seguro y eficaz en niños menores de 3 años.
¿Cuáles son los posibles efectos secundarios de Rebetol?
Rebetol puede causar efectos secundarios graves que incluyen:
- urticaria,
- respiración dificultosa,
- hinchazón de la cara, los labios, la lengua o la garganta,
- problemas de la vista
- dolor intenso en la parte superior del estómago que se extiende a la espalda,
- náuseas,
- vómitos,
- Diarrea,
- tos nueva o que empeora,
- fiebre,
- dolor punzante en el pecho,
- sibilancias,
- dificultad para respirar,
- depresión severa,
- pensamientos de autolesión,
- pensamientos de lastimar a otros,
- piel pálida o amarillenta,
- orina de color oscuro,
- confusión,
- debilidad,
- escalofríos,
- síntomas similares a la gripe,
- encías hinchadas,
- úlceras de boca,
- llagas en la piel,
- moretones con facilidad,
- sangrado inusual y
- aturdimiento
- Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
- Los efectos secundarios más comunes de Rebetol incluyen:
- náuseas,
- vómitos,
- pérdida de apetito,
- fiebre,
- escalofríos,
- sacudida,
- recuentos bajos de glóbulos,
- anemia,
- debilidad,
- cansancio,
- dolor de cabeza,
- dolor muscular,
- cambios de humor,
- ansiedad, y
- irritabilidad
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Rebetol. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
ADVERTENCIA
RIESGO DE TRASTORNOS GRAVES Y EFECTOS ASOCIADOS A LA RIBAVIRINA
- La monoterapia con REBETOL no es eficaz para el tratamiento de la infección crónica por el virus de la hepatitis C y no debe usarse solo para esta indicación [ver ADVERTENCIAS Y PRECAUCIONES].
- La principal toxicidad de la ribavirina es la anemia hemolítica. La anemia asociada con la terapia con REBETOL 200 mg puede provocar un empeoramiento de la enfermedad cardíaca que ha provocado infartos de miocardio fatales y no fatales. Los pacientes con antecedentes de enfermedad cardíaca importante o inestable no deben recibir tratamiento con REBETOL [ver DOSIS Y ADMINISTRACIÓN, ADVERTENCIAS Y PRECAUCIONES y REACCIONES ADVERSAS].
- Se han demostrado efectos teratogénicos y embriocidas significativos en todas las especies animales expuestas a la ribavirina. Además, la ribavirina tiene una vida media de dosis múltiples de 12 días, por lo que puede persistir en compartimentos no plasmáticos hasta por 6 meses. Por lo tanto, la terapia con REBETOL está contraindicada en mujeres embarazadas y en las parejas masculinas de mujeres embarazadas. Se debe tener sumo cuidado para evitar el embarazo durante la terapia y durante los 6 meses posteriores a la finalización del tratamiento tanto en pacientes femeninas como en las parejas femeninas de pacientes masculinos que reciben terapia con REBETOL. Se deben utilizar al menos dos formas confiables de anticoncepción efectiva durante el tratamiento y durante el período de seguimiento posterior al tratamiento de 6 meses [consulte CONTRAINDICACIONES, ADVERTENCIAS Y PRECAUCIONES, Uso en poblaciones específicas, Toxicología no clínica e INFORMACIÓN PARA EL PACIENTE].
DESCRIPCIÓN
REBETOL (ribavirina), es un análogo de nucleósido sintético (análogo de purina). El nombre químico de la ribavirina es 1-β-D-ribofuranosil-1H-1,2,4-triazol-3-carboxamida y tiene la siguiente fórmula estructural (ver Figura 1).
Figura 1: Fórmula estructural
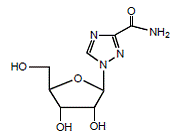
La ribavirina es un polvo cristalino blanco. Es libremente soluble en agua y ligeramente soluble en alcohol anhidro. La fórmula empírica es C8H12N4O5 y el peso molecular es 244,21.
Las cápsulas de REBETOL consisten en un polvo blanco en una cápsula de gelatina blanca y opaca. Cada cápsula contiene 200 mg de ribavirina y los ingredientes inactivos celulosa microcristalina, lactosa monohidrato, croscarmelosa sódica y estearato de magnesio. La cubierta de la cápsula consiste en gelatina, laurilsulfato de sodio, dióxido de silicio y dióxido de titanio. La cápsula está impresa con tinta farmacéutica azul comestible hecha de goma laca, alcohol etílico anhidro, alcohol isopropílico, alcohol n-butílico, propilenglicol, hidróxido de amonio y laca de aluminio FD&C Blue #2.
REBETOL 200 mg solución oral es un líquido transparente, de incoloro a amarillo pálido o amarillo claro con sabor a chicle. Cada mililitro de la solución contiene 40 mg de ribavirina y los ingredientes inactivos sacarosa, glicerina, sorbitol, propilenglicol, citrato de sodio, ácido cítrico, benzoato de sodio, sabor natural y artificial para chicle #15864 y agua.
INDICACIONES
Hepatitis C Crónica (CHC)
REBETOL® (ribavirina) en combinación con interferón alfa-2b (pegilado y no pegilado) está indicado para el tratamiento de la hepatitis C crónica (HCC) en pacientes de 3 años de edad y mayores con enfermedad hepática compensada [ver ADVERTENCIAS Y PRECAUCIONES , y Uso en poblaciones específicas ].
Se deben considerar los siguientes puntos al iniciar la terapia de combinación de REBETOL con PegIntron® o INTRON A®:
- Estas indicaciones se basan en lograr un ARN-VHC indetectable después del tratamiento durante 24 o 48 semanas y mantener una respuesta virológica sostenida (RVS) 24 semanas después de la última dosis.
- Se prefiere la terapia de combinación con REBETOL/PegIntron a REBETOL/INTRON A ya que esta combinación proporciona tasas de respuesta sustancialmente mejores [ver Estudios clínicos ].
- Los pacientes con las siguientes características tienen menos probabilidades de beneficiarse de un nuevo tratamiento después de un ciclo de tratamiento fallido: falta de respuesta previa, tratamiento previo con interferón pegilado, fibrosis en puente significativa o cirrosis e infección por genotipo 1 [ver Estudios clínicos ].
- No hay datos de seguridad y eficacia disponibles para tratamientos de más de un año.
DOSIFICACIÓN Y ADMINISTRACIÓN
Bajo ninguna circunstancia se deben abrir, triturar o romper las cápsulas de REBETOL. REBETOL 200 mg debe tomarse con alimentos [ver FARMACOLOGÍA CLÍNICA ]. REBETOL no debe usarse en pacientes con aclaramiento de creatinina inferior a 50 ml/min.
Terapia combinada de REBETOL/PegIntron
Pacientes adultos
La dosis recomendada de PegIntron es de 1,5 mcg/kg/semana por vía subcutánea en combinación con 800 a 1400 mg de REBETOL en cápsulas de 200 mg por vía oral según el peso corporal del paciente (consulte la Tabla 1). El volumen de PegIntron a inyectar depende de la concentración de PegIntron y del peso corporal del paciente; consulte la etiqueta de PegIntron para obtener información adicional sobre la dosificación.
Duración del tratamiento: pacientes sin tratamiento previo con interferón alfa
La duración del tratamiento para pacientes con genotipo 1 es de 48 semanas. Se debe considerar la interrupción de la terapia en pacientes que no logran una caída o pérdida de al menos 2 log10 del ARN-VHC a las 12 semanas, o si el ARN-VHC sigue siendo detectable después de 24 semanas de terapia. Los pacientes con genotipo 2 y 3 deben ser tratados durante 24 semanas.
Duración del tratamiento: retratamiento con PegIntron/REBETOL de los fracasos del tratamiento anterior
La duración del tratamiento para los pacientes que previamente fracasaron es de 48 semanas, independientemente del genotipo del VHC. Los pacientes tratados nuevamente que no logran un ARN-VHC indetectable en la semana 12 de la terapia, o cuyo ARN-VHC permanece detectable después de 24 semanas de terapia, es muy poco probable que logren una RVS y se debe considerar la interrupción de la terapia [ver Estudios clínicos ].
Pacientes pediátricos
La dosificación para pacientes pediátricos está determinada por el área de superficie corporal para PegIntron y por el peso corporal para REBETOL. La dosis recomendada de PegIntron es de 60 mcg/m²/semana por vía subcutánea en combinación con 15 mg/kg/día de REBETOL 200 mg por vía oral divididos en dos dosis (ver Tabla 2) para pacientes pediátricos de 3 a 17 años. Los pacientes que cumplen 18 años mientras reciben PegIntron/REBETOL 200 mg deben permanecer en el régimen de dosificación pediátrico. La duración del tratamiento para pacientes con genotipo 1 es de 48 semanas. Los pacientes con genotipo 2 y 3 deben ser tratados durante 24 semanas.
Tratamiento combinado con REBETOL/INTRON A
Adultos
Duración del tratamiento: pacientes sin tratamiento previo con interferón alfa
La dosis recomendada de INTRON A es de 3 millones de UI tres veces por semana por vía subcutánea. La dosis recomendada de cápsulas de REBETOL depende del peso corporal del paciente (consulte la Tabla 3). La duración recomendada del tratamiento para pacientes no tratados previamente con interferón es de 24 a 48 semanas. La duración del tratamiento debe ser individualizada para el paciente dependiendo de las características basales de la enfermedad, la respuesta a la terapia y la tolerabilidad del régimen [ver INDICACIONES Y USO , REACCIONES ADVERSAS , y Estudios clínicos ]. Después de 24 semanas de tratamiento, se debe evaluar la respuesta virológica. Se debe considerar la interrupción del tratamiento en cualquier paciente que no haya alcanzado un ARN-VHC por debajo del límite de detección del ensayo a las 24 semanas. No hay datos de seguridad y eficacia sobre el tratamiento durante más de 48 semanas en la población de pacientes no tratados previamente.
Duración del tratamiento: retratamiento con INTRON A/REBETOL en pacientes con recaída
En pacientes que recaen después de la monoterapia con interferón no pegilado, la duración recomendada del tratamiento es de 24 semanas.
Pediatría
La dosis recomendada de REBETOL es de 15 mg/kg por día por vía oral (dosis dividida por la mañana y por la noche). Consulte la Tabla 2 para ver la dosificación pediátrica de REBETOL en combinación con INTRON A. INTRON A para inyección por peso corporal de 25 kg a 61 kg es de 3 millones de UI/m² tres veces por semana por vía subcutánea. Consulte la tabla de dosificación para adultos para más de 61 kg de peso corporal.
La duración recomendada del tratamiento es de 48 semanas para pacientes pediátricos con genotipo 1. Después de 24 semanas de tratamiento, se debe evaluar la respuesta virológica. Se debe considerar la interrupción del tratamiento en cualquier paciente que no haya alcanzado un ARN-VHC por debajo del límite de detección del ensayo en este momento. La duración recomendada del tratamiento para pacientes pediátricos con genotipo 2/3 es de 24 semanas.
Pruebas de laboratorio
Se recomiendan las siguientes pruebas de laboratorio para todos los pacientes tratados con REBETOL 200 mg, antes de comenzar el tratamiento y luego periódicamente.
Pruebas hematológicas estándar, incluida la hemoglobina (pretratamiento, Semana 2 y Semana 4 de terapia, y según corresponda clínicamente [ver ADVERTENCIAS Y PRECAUCIONES ], recuentos completos y diferenciales de glóbulos blancos y recuento de plaquetas.
- Química sanguínea: pruebas de función hepática y TSH.
- Embarazo: incluido el control mensual de las mujeres en edad fértil.
- ECG [ver ADVERTENCIAS Y PRECAUCIONES ].
Modificaciones de dosis
Si se desarrollan reacciones adversas graves o anomalías de laboratorio durante el tratamiento combinado con REBETOL/INTRON A o REBETOL/PegIntron, modifique o interrumpa la dosis hasta que la reacción adversa disminuya o disminuya en gravedad [ver ADVERTENCIAS Y PRECAUCIONES ]. Si la intolerancia persiste después del ajuste de la dosis, se debe interrumpir el tratamiento combinado. La reducción de la dosis de PegIntron en pacientes adultos que reciben tratamiento combinado con REBETOL/PegIntron se logra en un proceso de dos pasos desde la dosis inicial original de 1,5 mcg/kg/semana, a 1 mcg/kg/semana y luego a 0,5 mcg/kg/semana. , si es necesario. Consulte la etiqueta de PegIntron para obtener información adicional sobre la reducción de la dosis de PegIntron.
En el Estudio 2 de terapia combinada para adultos, se produjeron reducciones de dosis en el 42 % de los sujetos que recibieron PegIntron 1,5 mcg/kg y REBETOL 800 mg al día, incluido el 57 % de los sujetos que pesaban 60 kg o menos. En el Estudio 4, al 16 % de los sujetos se les redujo la dosis de PegIntron a 1 mcg/kg en combinación con REBETOL, y un 4 % adicional requirió la segunda reducción de la dosis de PegIntron a 0,5 mcg/kg debido a eventos adversos [ver REACCIONES ADVERSAS ].
La reducción de la dosis en pacientes pediátricos se logra modificando la dosis recomendada de PegIntron en un proceso de dos pasos desde la dosis inicial original de 60 mcg/m²/semana a 40 mcg/m²/semana y luego a 20 mcg/m²/semana, si necesarios (ver Tabla 4). En el ensayo de terapia combinada pediátrica, se produjeron reducciones de dosis en el 25 % de los sujetos que recibieron 60 mcg/m² de PegIntron semanalmente y 15 mg/kg de REBETOL al día. La reducción de la dosis en pacientes pediátricos se logra modificando la dosis recomendada de REBETOL de la dosis inicial original de 15 mg/kg al día en un proceso de dos pasos a 12 mg/kg/día y luego a 8 mg/kg/día, si es necesario ( ver Tabla 4).
REBETOL 200 mg no debe usarse en pacientes con aclaramiento de creatinina inferior a 50 ml/min. Los pacientes con función renal alterada y los mayores de 50 años deben ser monitoreados cuidadosamente con respecto al desarrollo de anemia [ver ADVERTENCIAS Y PRECAUCIONES , Uso en poblaciones específicas , y FARMACOLOGÍA CLÍNICA ].
REBETOL 200 mg debe administrarse con precaución a pacientes con enfermedad cardíaca preexistente. Los pacientes deben ser evaluados antes de comenzar la terapia y deben ser monitoreados apropiadamente durante la terapia. Si hay algún deterioro del estado cardiovascular, se debe suspender la terapia [ver ADVERTENCIAS Y PRECAUCIONES ].
Para pacientes con antecedentes de enfermedad cardiovascular estable, se requiere una reducción permanente de la dosis si la hemoglobina disminuye en 2 g/dL o más durante cualquier período de 4 semanas. Además, para estos pacientes con antecedentes cardíacos, si la hemoglobina permanece por debajo de 12 g/dl después de 4 semanas con una dosis reducida, el paciente debe suspender la terapia combinada.
Se recomienda que un paciente cuyo nivel de hemoglobina caiga por debajo de 10 g/dl modifique o interrumpa su dosis de REBETOL según la Tabla 4 [ver ADVERTENCIAS Y PRECAUCIONES ].
Consulte la etiqueta de INTRON A o PegIntron para obtener información adicional sobre cómo reducir una dosis de INTRON A o PegIntron.
Interrupción de la dosificación
Adultos
En el genotipo 1 del VHC, los pacientes sin tratamiento previo con interferón alfa que reciben PegIntron en combinación con ribavirina, se recomienda la interrupción del tratamiento si no hay una caída o pérdida de al menos 2 log10 del ARN-VHC a las 12 semanas de tratamiento, o si el ARN-VHC los niveles siguen siendo detectables después de 24 semanas de tratamiento. Independientemente del genotipo, es muy poco probable que los pacientes tratados previamente que tienen ARN-VHC detectable en la semana 12 o 24 alcancen la RVS y se debe considerar la interrupción del tratamiento.
Pediatría (3-17 años de edad)
Se recomienda que los pacientes que reciben la combinación de PegIntron/REBETOL (excluyendo los genotipos 2 y 3 del VHC) interrumpan el tratamiento a las 12 semanas si su tratamiento en la semana 12 del ARN-VHC disminuyó menos de 2 log10 en comparación con un pretratamiento o a las 24 semanas si tienen síntomas detectables. ARN-VHC en la semana 24 de tratamiento.
CÓMO SUMINISTRADO
Formas de dosificación y concentraciones
REBETOL 200mg Cápsulas 200mg
REBETOL 200mg Solución Oral 40 mg por mL
Almacenamiento y manipulación
REBETOL 200 mg Cápsulas son cápsulas opacas de color blanco con REBETOL, 200 mg, y el logotipo de Schering Corporation impreso en la cubierta de la cápsula; las cápsulas están envasadas en un frasco que contiene 56 cápsulas ( CDN 0085-1351-05), 70 cápsulas ( CDN 0085-1385-07), y 84 cápsulas ( CDN 0085-1194-03).
REBETOL 200mg Solución Oral 40mg por ml es un líquido transparente, de incoloro a amarillo pálido o claro con sabor a chicle y se envasa en botellas de vidrio ámbar de 4 oz (100 ml/botella) con cierres a prueba de niños ( CDN 0085-1318-01).
El frasco de Cápsulas de REBETOL debe almacenarse a 25°C (77°F); excursiones permitidas a 15-30°C (59-86°F) [ver Temperatura ambiente controlada por USP ].
La solución oral de 200 mg de REBETOL debe almacenarse entre 2 y 8 °C (36 y 46 °F) o a 25 °C (77 °F); excursiones permitidas a 15-30°C (59-86°F) [ver Temperatura ambiente controlada por USP ].
REBETOL 200 mg Solución oral fabricado para: Merck Sharp & Dohme Corp., una subsidiaria de Whitehouse Station, NJ 08889, EE. UU. Fabricado por: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canadá Cápsulas REBETOL fabricadas por: Merck Sharp & Dohme Corp., una subsidiaria de Whitehouse Station, NJ 08889, EE. UU. Revisado: diciembre de 2014.EFECTOS SECUNDARIOS
Se han realizado ensayos clínicos con REBETOL en combinación con PegIntron o INTRON A en más de 7800 sujetos de 3 a 76 años de edad.
La principal toxicidad de la ribavirina es la anemia hemolítica. Las reducciones en los niveles de hemoglobina ocurrieron dentro de las primeras 1 a 2 semanas de terapia oral. Se produjeron reacciones cardíacas y pulmonares asociadas con anemia en aproximadamente el 10 % de los pacientes [ver ADVERTENCIAS Y PRECAUCIONES ].
Más del 96% de todos los sujetos en ensayos clínicos experimentaron una o más reacciones adversas. Las reacciones adversas notificadas con mayor frecuencia en sujetos adultos que recibieron PegIntron o INTRON A en combinación con REBETOL 200 mg fueron inflamación/reacción en el lugar de la inyección, fatiga/astenia, dolor de cabeza, escalofríos, fiebre, náuseas, mialgia y ansiedad/labilidad emocional/irritabilidad. Las reacciones adversas más comunes en sujetos pediátricos, de 3 años en adelante, que recibieron REBETOL 200 mg en combinación con PegIntron o INTRON A fueron pirexia, dolor de cabeza, neutropenia, fatiga, anorexia, eritema en el lugar de la inyección y vómitos.
La sección de Reacciones adversas hace referencia a los siguientes ensayos clínicos:
- Ensayos de terapia combinada REBETOL/PegIntron:
- Estudio clínico 1: evaluó la monoterapia con PegIntron (no se describe con más detalle en esta etiqueta; consulte la etiqueta de PegIntron para obtener información sobre este ensayo).
- Estudio 2: evaluó la dosis fija de REBETOL de 800 mg/día en combinación con 1,5 mcg/kg/semana de PegIntron o con INTRON A.
- Estudio 3: evaluó PegIntron/REBETOL basado en el peso de 200 mg en combinación con PegIntron/régimen de dosis plana de REBETOL de 200 mg.
- Estudio 4: comparó dos dosis de PegIntron (1,5 mcg/kg/semana y 1 mcg/kg/semana) en combinación con REBETOL y un tercer grupo de tratamiento que recibió Pegasys® (180 mcg/semana)/Copegus® (1000-1200 mg/día) ).
- Estudio 5: evaluó PegIntron (1,5 mcg/kg/semana) en combinación con REBETOL basado en el peso en sujetos con fracaso previo del tratamiento.
- Terapia combinada de PegIntron/REBETOL 200 mg en pacientes pediátricos
- Ensayos de terapia combinada REBETOL/INTRON A para adultos y pediatría
Se han producido reacciones adversas graves en aproximadamente el 12 % de los sujetos en ensayos clínicos con PegIntron con o sin REBETOL [ver ADVERTENCIA EN CAJA , ADVERTENCIAS Y PRECAUCIONES ]. Los eventos graves más comunes que ocurrieron en sujetos tratados con PegIntron y REBETOL 200 mg fueron depresión e ideación suicida [ver ADVERTENCIAS Y PRECAUCIONES ], cada uno ocurriendo a una frecuencia de menos del 1%. La ideación o los intentos de suicidio ocurrieron con mayor frecuencia entre los pacientes pediátricos, principalmente adolescentes, en comparación con los pacientes adultos (2,4 % frente al 1 %) durante el tratamiento y el seguimiento sin tratamiento [ver ADVERTENCIAS Y PRECAUCIONES ]. La reacción fatal más común que ocurrió en sujetos tratados con PegIntron y REBETOL 200 mg fue paro cardíaco, ideación suicida e intento de suicidio [ver ADVERTENCIAS Y PRECAUCIONES ], todos ocurriendo en menos del 1% de los sujetos.
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica clínica.
Experiencia en ensayos clínicos: terapia combinada de REBETOL/PegIntron
Sujetos Adultos
Las reacciones adversas que ocurrieron en el ensayo clínico con una incidencia superior al 5 % se proporcionan por grupo de tratamiento del tratamiento combinado de REBETOL/PegIntron (Estudio 2) en la Tabla 5.
La Tabla 6 resume las reacciones adversas relacionadas con el tratamiento en el Estudio 4 que ocurrieron con una incidencia mayor o igual al 10 %.
La incidencia de reacciones adversas graves fue comparable en todos los ensayos. En el Estudio 3, se informó una incidencia similar de reacciones adversas graves para el grupo de REBETOL basado en el peso (12 %) y para el régimen de dosis fija de REBETOL. En el Estudio 2, la incidencia de reacciones adversas graves fue del 17 % en los grupos de PegIntron/REBETOL en comparación con el 14 % en el grupo de INTRON A/REBETOL.
En muchos, pero no en todos los casos, las reacciones adversas se resolvieron después de la reducción de la dosis o la interrupción del tratamiento. Algunos sujetos experimentaron reacciones adversas graves en curso o nuevas durante el período de seguimiento de 6 meses. En el Estudio 2, muchos sujetos continuaron experimentando reacciones adversas varios meses después de suspender la terapia. Al final del período de seguimiento de 6 meses, la incidencia de reacciones adversas en curso por clase corporal en el grupo de 200 mg de PegIntron 1.5/REBETOL fue del 33 % (psiquiátrico), del 20 % (musculoesquelético) y del 10 % (para trastornos endocrinos y para IG). En aproximadamente 10 a 15% de los sujetos, la pérdida de peso, la fatiga y el dolor de cabeza no se resolvieron.
Ha habido 31 muertes de sujetos que ocurrieron durante el tratamiento o durante el seguimiento en estos ensayos clínicos. En el Estudio 1, hubo 1 suicidio en un sujeto que recibió la monoterapia con PegIntron y 2 muertes entre los sujetos que recibieron la monoterapia con INTRON A (1 asesinato/suicidio y 1 muerte súbita). En el Estudio 2, hubo 1 suicidio en un sujeto que recibió la terapia combinada de PegIntron/REBETOL 200 mg; y la muerte de 1 sujeto en el grupo de 200 mg de INTRON A/REBETOL (accidente automovilístico). En el Estudio 3, hubo 14 muertes, 2 de las cuales fueron suicidios probables y 1 fue una muerte inexplicable en una persona con antecedentes médicos relevantes de depresión. En el Estudio 4, hubo 12 muertes, 6 de las cuales ocurrieron en sujetos que recibieron la terapia combinada de PegIntron/REBETOL, 5 en el brazo de PegIntron 1.5 mcg/REBETOL (N=1019) y 1 en el brazo de PegIntron 1 mcg/REBETOL (N= 1016), y 6 de los cuales ocurrieron en sujetos que recibieron Pegasys/Copegus (N=1035); hubo 3 suicidios que ocurrieron durante el período de seguimiento sin tratamiento en sujetos que recibieron la terapia combinada de PegIntron (1.5 mcg/kg)/REBETOL 200 mg.
En los Estudios 1 y 2, del 10 al 14 % de los sujetos que recibieron PegIntron, solo o en combinación con REBETOL 200 mg, interrumpieron el tratamiento en comparación con el 6 % tratado con INTRON A solo y el 13 % tratado con INTRON A en combinación con REBETOL. De manera similar, en el Estudio 3, el 15 % de los sujetos que recibieron PegIntron en combinación con REBETOL basado en el peso y el 14 % de los sujetos que recibieron PegIntron y una dosis fija de 200 mg de REBETOL interrumpieron el tratamiento debido a una reacción adversa. Las razones más comunes para la interrupción de la terapia estaban relacionadas con los efectos conocidos del interferón de reacciones adversas psiquiátricas, sistémicas (p. ej., fatiga, dolor de cabeza) o gastrointestinales. En el Estudio 4, el 13 % de los sujetos en el brazo de PegIntron 1,5 mcg/REBETOL 200 mg, el 10 % en el brazo de PegIntron 1 mcg/REBETOL 200 mg y el 13 % en el brazo de Pegasys 180 mcg/Copegus interrumpieron el tratamiento debido a eventos adversos.
En el Estudio 2, se produjeron reducciones de dosis debido a reacciones adversas en el 42 % de los sujetos que recibieron PegIntron (1,5 mcg/kg)/200 mg de REBETOL y en el 34 % de los que recibieron INTRON A/REBETOL. La mayoría de los sujetos (57 %) con un peso de 60 kg o menos que recibieron PegIntron (1,5 mcg/kg)/REBETOL requirieron una reducción de la dosis. La reducción de interferón estuvo relacionada con la dosis (PegIntron 1,5 mcg/kg mayor que PegIntron 0,5 mcg/kg o INTRON A), 40%, 27%, 28%, respectivamente. La reducción de la dosis de REBETOL 200 mg fue similar en los tres grupos, del 33 al 35 %. Las razones más comunes para modificar la dosis fueron neutropenia (18%) o anemia (9%) (ver Valores de laboratorio ). Otras razones comunes incluyeron depresión, fatiga, náuseas y trombocitopenia. En el Estudio 3, las modificaciones de dosis debido a reacciones adversas ocurrieron con mayor frecuencia con la dosificación basada en el peso (WBD) en comparación con la dosificación plana (29 % y 23 %, respectivamente). En el Estudio 4, el 16 % de los sujetos tuvo una reducción de la dosis de PegIntron a 1 mcg/kg en combinación con REBETOL 200 mg, y un 4 % adicional requirió la segunda reducción de la dosis de PegIntron a 0,5 mcg/kg debido a eventos adversos en comparación con el 15 % de sujetos en el brazo de Pegasys/Copegus, que requirieron una reducción de la dosis a 135 mcg/semana con Pegasys, con un 7 % adicional en el brazo de Pegasys/Copegus que requirió una segunda reducción de la dosis a 90 mcg/semana con Pegasys.
En los ensayos de combinación de PegIntron/REBETOL de 200 mg, las reacciones adversas más comunes fueron psiquiátricas, que ocurrieron entre el 77 % de los sujetos del Estudio 2 y entre el 68 % y el 69 % de los sujetos del Estudio 3. Estas reacciones adversas psiquiátricas incluyeron con mayor frecuencia depresión, irritabilidad y insomnio, cada uno informado por aproximadamente 30% a 40% de los sujetos en todos los grupos de tratamiento. El comportamiento suicida (ideación, intentos y suicidios) ocurrió en el 2% de todos los sujetos durante el tratamiento o durante el seguimiento después de la interrupción del tratamiento [ver ADVERTENCIAS Y PRECAUCIONES ]. En el Estudio 4, se produjeron reacciones adversas psiquiátricas en el 58 % de los sujetos del brazo de PegIntron 1,5 mcg/REBETOL 200 mg, el 55 % de los sujetos en el brazo de PegIntron 1 mcg/REBETOL 200 mg y el 57 % de los sujetos en el brazo de Pegasys 180 mcg/Copegus .
PegIntron indujo fatiga o dolor de cabeza en aproximadamente dos tercios de los sujetos, con fiebre o escalofríos en aproximadamente la mitad de los sujetos. La gravedad de algunos de estos síntomas sistémicos (p. ej., fiebre y dolor de cabeza) tendió a disminuir a medida que continuaba el tratamiento. En los Estudios 1 y 2, la inflamación y la reacción en el lugar de la aplicación (p. ej., hematomas, picazón e irritación) se produjeron con aproximadamente el doble de incidencia con las terapias de PegIntron (en hasta el 75 % de los sujetos) en comparación con INTRON A. Sin embargo, el dolor en el lugar de la inyección fue menor. infrecuente (2 a 3%) en todos los grupos. En el Estudio 3, hubo una incidencia general del 23 % al 24 % de reacciones o inflamación en el lugar de la inyección.
Los sujetos que recibieron REBETOL/PegIntron como retratamiento después del fracaso de un régimen previo de combinación de interferón informaron reacciones adversas similares a las asociadas anteriormente con este régimen durante los ensayos clínicos de sujetos sin tratamiento previo.
Sujetos pediátricos
En general, el perfil de reacciones adversas en la población pediátrica fue similar al observado en adultos. En el ensayo pediátrico, las reacciones adversas más prevalentes en todos los sujetos fueron pirexia (80 %), dolor de cabeza (62 %), neutropenia (33 %), fatiga (30 %), anorexia (29 %), eritema en el lugar de la inyección (29 %). %) y vómitos (27%). La mayoría de las reacciones adversas notificadas en el ensayo fueron de gravedad leve o moderada. Se informaron reacciones adversas graves en el 7 % (8/107) de todos los sujetos e incluyeron dolor en el lugar de la inyección (1 %), dolor en las extremidades (1 %), dolor de cabeza (1 %), neutropenia (1 %) y pirexia (4 %). Las reacciones adversas importantes que ocurrieron en esta población de sujetos fueron nerviosismo (7%; 7/107), agresión (3%; 3/107), ira (2%; 2/107) y depresión (1%; 1/107) . Cinco sujetos recibieron tratamiento con levotiroxina, tres con hipotiroidismo clínico y dos con elevaciones asintomáticas de TSH. El aumento de peso y altura de los sujetos pediátricos tratados con PegIntron más REBETOL fue inferior al previsto por los datos normativos de la población durante todo el tratamiento. Se observó una velocidad de crecimiento gravemente inhibida (menos del percentil 3) en el 70 % de los sujetos durante el tratamiento.
Se requirieron modificaciones de dosis de PegIntron y/o ribavirina en el 25 % de los sujetos debido a reacciones adversas relacionadas con el tratamiento, más comúnmente anemia, neutropenia y pérdida de peso. Dos sujetos (2 %; 2/107) interrumpieron el tratamiento como resultado de una reacción adversa.
Las reacciones adversas que ocurrieron con una incidencia mayor o igual al 10 % en los sujetos del ensayo pediátrico se proporcionan en la Tabla 7.
Noventa y cuatro de 107 sujetos se inscribieron en un ensayo de seguimiento a largo plazo de 5 años. Los efectos a largo plazo sobre el crecimiento fueron menores en los sujetos tratados durante 24 semanas que en los tratados durante 48 semanas. El veinticuatro por ciento de los sujetos (11/46) tratados durante 24 semanas y el 40 % de los sujetos (19/48) tratados durante 48 semanas tuvieron una disminución de la altura para la edad > percentil 15 desde el pretratamiento hasta el final de los 5 años. seguimiento a largo plazo en comparación con los percentiles iniciales previos al tratamiento. Se observó que el 11 % de los sujetos (5/46) tratados durante 24 semanas y el 13 % de los sujetos (6/48) tratados durante 48 semanas tuvieron una disminución desde el inicio previo al tratamiento de > 30 percentiles de altura para la edad hasta el final del seguimiento a largo plazo de 5 años. Si bien se observó en todos los grupos de edad, el mayor riesgo de reducción de la estatura al final del seguimiento a largo plazo pareció correlacionarse con el inicio de la terapia combinada durante los años de velocidad máxima de crecimiento esperada. [Ver ADVERTENCIAS Y PRECAUCIONES ]
Valores de laboratorio
Sujetos adultos y pediátricos
El perfil de reacciones adversas en el Estudio 3, que comparó la combinación de PegIntron/REBETOL de 200 mg según el peso con un régimen de PegIntron/dosis plana de REBETOL de 200 mg, reveló un aumento en la tasa de anemia con la dosificación según el peso (29 % frente a 19 % para la administración según el peso). vs. regímenes de dosis plana, respectivamente). Sin embargo, la mayoría de los casos de anemia fueron leves y respondieron a las reducciones de dosis.
A continuación se describen los cambios en valores de laboratorio seleccionados durante el tratamiento en combinación con REBETOL 200 mg. Las disminuciones de hemoglobina, leucocitos, neutrófilos y plaquetas pueden requerir una reducción de la dosis o la suspensión permanente del tratamiento. [ver DOSIFICACIÓN Y ADMINISTRACIÓN ]. Los cambios en los valores de laboratorio seleccionados durante la terapia se describen en la Tabla 8. La mayoría de los cambios en los valores de laboratorio en el ensayo de PegIntron/REBETOL 200 mg con pediatría fueron leves o moderados.
Hemoglobina
Los niveles de hemoglobina disminuyeron a menos de 11 g/dL en aproximadamente el 30 % de los sujetos del Estudio 2. En el Estudio 3, el 47 % de los sujetos que recibieron WBD REBETOL 200 mg y el 33 % de los sujetos que recibieron una dosis fija de REBETOL 200 mg tuvieron reducciones en los niveles de hemoglobina de menos de 11 g /dl. Las reducciones en la hemoglobina a menos de 9 g/dL ocurrieron con mayor frecuencia en sujetos que recibieron WBD en comparación con la dosis plana (4 % y 2 %, respectivamente). En el Estudio 2, se requirió modificar la dosis en el 9 % y el 13 % de los sujetos en los grupos de PegIntron/REBETOL 200 mg e INTRON A/REBETOL 200 mg. En el Estudio 4, los sujetos que recibieron PegIntron (1,5 mcg/kg)/REBETOL tuvieron disminuciones en los niveles de hemoglobina entre 8,5 y menos de 10 g/dL (28 %) y menos de 8,5 g/dL (3 %), mientras que en los pacientes recibiendo Pegasys 180 mcg/Copegus estas disminuciones ocurrieron en 26% y 4% de los sujetos respectivamente. Los niveles de hemoglobina se estabilizaron en las semanas de tratamiento 4-6 en promedio. El patrón típico observado fue una disminución en los niveles de hemoglobina en la semana 4 del tratamiento, seguida de una estabilización y una meseta, que se mantuvo hasta el final del tratamiento. En el ensayo de monoterapia con PegIntron, las disminuciones de hemoglobina fueron generalmente leves y rara vez fue necesario modificar la dosis [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
neutrófilos
Se observaron disminuciones en los recuentos de neutrófilos en la mayoría de los sujetos adultos tratados con terapia combinada con REBETOL 200 mg en el Estudio 2 (85 %) e INTRON A/REBETOL (60 %). Se produjo neutropenia grave potencialmente mortal (menos de 0,5 x 109/L) en el 2 % de los sujetos tratados con INTRON A/REBETOL 200 mg y en aproximadamente el 4 % de los sujetos tratados con PegIntron/REBETOL 200 mg en el Estudio 2. El dieciocho por ciento de los sujetos que recibieron PegIntron/REBETOL en el Estudio 2 requirió la modificación de la dosis de interferón. Pocos sujetos (menos del 1%) requirieron la interrupción permanente del tratamiento. Los recuentos de neutrófilos generalmente regresaron a los niveles previos al tratamiento 4 semanas después de la interrupción de la terapia [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
plaquetas
Los recuentos de plaquetas disminuyeron a menos de 100 000/mm³ en aproximadamente el 20 % de los sujetos tratados con PegIntron solo o con 200 mg de REBETOL y en el 6 % de los sujetos adultos tratados con INTRON A/REBETOL. Disminuciones graves en el recuento de plaquetas (menos de 50 000/mm³) ocurren en menos del 4 % de los sujetos adultos. Los pacientes pueden requerir la interrupción o la modificación de la dosis como resultado de la disminución de plaquetas [ver DOSIFICACIÓN Y ADMINISTRACIÓN ]. En el Estudio 2, el 1 % o el 3 % de los sujetos requirieron una modificación de la dosis de INTRON A o PegIntron, respectivamente. Los recuentos de plaquetas generalmente volvieron a los niveles previos al tratamiento 4 semanas después de la interrupción de la terapia.
función tiroidea
El desarrollo de anormalidades de TSH, con o sin manifestaciones clínicas, está asociado con las terapias con interferón. En el Estudio 2, se produjeron trastornos tiroideos clínicamente evidentes entre sujetos tratados con INTRON A o PegIntron (con o sin REBETOL) con una incidencia similar (5 % para hipotiroidismo y 3 % para hipertiroidismo). Los sujetos desarrollaron anomalías de TSH de nueva aparición durante el tratamiento y durante el período de seguimiento. Al final del período de seguimiento, el 7% de los sujetos todavía tenían valores anormales de TSH.
Bilirrubina y ácido úrico
En el Estudio 2, del 10 al 14 % de los sujetos desarrollaron hiperbilirrubinemia y del 33 al 38 % desarrollaron hiperuricemia asociada con hemólisis. Seis sujetos desarrollaron gota de leve a moderada.
Experiencia en ensayos clínicos: terapia combinada REBETOL/INTRON A
Sujetos Adultos
En los ensayos clínicos, el 19 % y el 6 % de los sujetos que no recibieron tratamiento previo y los que recayeron, respectivamente, interrumpieron el tratamiento debido a reacciones adversas en los brazos de combinación en comparación con el 13 % y el 3 % en los brazos de interferón. Las reacciones adversas seleccionadas relacionadas con el tratamiento que ocurrieron en los ensayos de EE. UU. con una incidencia mayor o igual al 5 % se proporcionan por grupo de tratamiento (consulte la Tabla 9). En general, las reacciones adversas relacionadas con el tratamiento seleccionadas se informaron con menor incidencia en los ensayos internacionales en comparación con los ensayos estadounidenses, con la excepción de astenia, síntomas similares a los de la influenza, nerviosismo y prurito.
Sujetos pediátricos
En ensayos clínicos de 118 sujetos pediátricos de 3 a 16 años de edad, el 6 % interrumpió el tratamiento debido a reacciones adversas. Se requirieron modificaciones de dosis en el 30% de los sujetos, más comúnmente por anemia y neutropenia. En general, el perfil de reacciones adversas en la población pediátrica fue similar al observado en adultos. Los trastornos en el lugar de la inyección, fiebre, anorexia, vómitos y labilidad emocional ocurrieron con mayor frecuencia en sujetos pediátricos en comparación con sujetos adultos. Por el contrario, los sujetos pediátricos experimentaron menos fatiga, dispepsia, artralgia, insomnio, irritabilidad, problemas de concentración, disnea y prurito en comparación con los sujetos adultos. Las reacciones adversas seleccionadas relacionadas con el tratamiento que ocurrieron con una incidencia mayor o igual al 5 % entre todos los sujetos pediátricos que recibieron la dosis recomendada de terapia combinada de REBETOL/INTRON A se proporcionan en la Tabla 9.
Durante un curso de terapia de 48 semanas, hubo una disminución en la tasa de crecimiento lineal (disminución de la asignación de percentil promedio del 7 %) y una disminución en la tasa de aumento de peso (disminución de la asignación de percentil promedio del 9 %). Se observó una inversión general de estas tendencias durante el período posterior al tratamiento de 24 semanas. Sin embargo, los datos a largo plazo en un número limitado de pacientes sugieren que la terapia combinada puede inducir una inhibición del crecimiento que resulta en una reducción de la estatura adulta final en algunos pacientes [ver ADVERTENCIAS Y PRECAUCIONES ].
Valores de laboratorio
Los cambios en valores hematológicos seleccionados (hemoglobina, glóbulos blancos, neutrófilos y plaquetas) durante la terapia se describen a continuación (consulte la Tabla 10).
Hemoglobina. Las disminuciones de hemoglobina entre los sujetos que recibieron la terapia con REBETOL comenzaron en la semana 1 y se estabilizaron en la semana 4. En sujetos no tratados previamente tratados durante 48 semanas, la disminución máxima media desde el inicio fue de 3,1 g/dl en el ensayo de EE. UU. y de 2,9 g/dl en el ensayo internacional. prueba. En los sujetos con recaída, la disminución máxima media desde el inicio fue de 2,8 g/dL en el ensayo de EE. UU. y de 2,6 g/dL en el ensayo internacional. Los valores de hemoglobina volvieron a los niveles previos al tratamiento dentro de las 4 a 8 semanas posteriores al cese de la terapia en la mayoría de los sujetos.
Bilirrubina y ácido úrico. En los ensayos clínicos se observaron aumentos tanto de la bilirrubina como del ácido úrico, asociados con la hemólisis. La mayoría fueron cambios bioquímicos moderados y se revirtieron dentro de las 4 semanas posteriores a la interrupción del tratamiento. Esta observación ocurrió con mayor frecuencia en sujetos con un diagnóstico previo de síndrome de Gilbert. Esto no se ha asociado con disfunción hepática o morbilidad clínica.
Experiencias posteriores a la comercialización
Se han identificado y notificado las siguientes reacciones adversas durante el uso posterior a la aprobación de REBETOL en combinación con INTRON A o PegIntron. Debido a que estas reacciones son informadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos de la sangre y del sistema linfático
Aplasia pura de glóbulos rojos, anemia aplásica
Trastornos del oído y del laberinto
Trastorno auditivo, vértigo
Trastornos respiratorios, torácicos y mediastínicos
Hipertensión pulmonar
Trastornos oculares
Desprendimiento de retina seroso
Desordenes endocrinos
Diabetes
INTERACCIONES CON LA DROGAS
didanosina
La exposición a didanosina oa su metabolito activo (didesoxiadenosina 5'-trifosfato) aumenta cuando se coadministra didanosina con ribavirina, lo que podría causar o empeorar toxicidades clínicas; por lo tanto, está contraindicada la coadministración de REBETOL 200 mg en cápsulas o solución oral y didanosina. En los ensayos clínicos se han notificado casos de insuficiencia hepática mortal, así como de neuropatía periférica, pancreatitis e hiperlactatemia/acidosis láctica sintomática.
Análogos de nucleósidos
Se ha producido descompensación hepática (algunas fatales) en pacientes cirróticos coinfectados por VIH/VHC que reciben terapia antirretroviral combinada para VIH e interferón alfa y ribavirina. Agregar tratamiento con interferones alfa solos o en combinación con ribavirina puede aumentar el riesgo en esta población de pacientes. Los pacientes que reciben interferón con ribavirina e inhibidores nucleósidos de la transcriptasa inversa (NRTI) deben ser monitoreados de cerca por toxicidades asociadas al tratamiento, especialmente descompensación hepática y anemia. La interrupción de los NRTI se debe considerar como médicamente apropiada (ver etiquetado para productos NRTI individuales ). También se debe considerar la reducción de la dosis o la interrupción del interferón, la ribavirina o ambos si se observa un empeoramiento de las toxicidades clínicas, incluida la descompensación hepática (p. ej., Child-Pugh superior a 6).
La ribavirina puede antagonizar la actividad antiviral del cultivo celular de la estavudina y la zidovudina contra el VIH. Se ha demostrado que la ribavirina en cultivos celulares inhibe la fosforilación de lamivudina, estavudina y zidovudina, lo que podría conducir a una disminución de la actividad antirretroviral. Sin embargo, en un estudio con otro interferón pegilado en combinación con ribavirina, no se observó interacción farmacocinética (p. ej., concentraciones plasmáticas o concentraciones de metabolitos activos trifosforilados intracelulares) ni farmacodinámica (p. ej., pérdida de la supresión virológica del VIH/VHC) cuando ribavirina y lamivudina (n =18), se coadministraron estavudina (n=10) o zidovudina (n=6) como parte de un régimen de múltiples fármacos en sujetos coinfectados por VIH/VHC. Por lo tanto, el uso concomitante de ribavirina con cualquiera de estos medicamentos debe usarse con precaución.
Fármacos metabolizados por el citocromo P-450
Los resultados de los estudios in vitro con preparaciones de microsomas hepáticos humanos y de rata indicaron poco o ningún metabolismo de la ribavirina mediado por la enzima citocromo P-450, con un potencial mínimo de interacciones farmacológicas basadas en la enzima P-450.
No se observaron interacciones farmacocinéticas entre INTRON A y las cápsulas de 200 mg de REBETOL en un estudio farmacocinético de dosis múltiples.
azatioprina
Se ha informado que el uso de ribavirina para el tratamiento de la hepatitis C crónica en pacientes que reciben azatioprina induce pancitopenia grave y puede aumentar el riesgo de mielotoxicidad relacionada con azatioprina. La inosina monofosfato deshidrogenasa (IMDH) es necesaria para una de las vías metabólicas de la azatioprina. Se sabe que la ribavirina inhibe la IMDH, lo que conduce a la acumulación de un metabolito de azatioprina, el monofosfato de 6-metiltioinosina (6-MTITP), que se asocia con mielotoxicidad (neutropenia, trombocitopenia y anemia). Los pacientes que reciben azatioprina con ribavirina deben tener hemogramas completos, incluidos los recuentos de plaquetas, monitoreados semanalmente durante el primer mes, dos veces al mes durante el segundo y tercer mes de tratamiento, luego mensualmente o con mayor frecuencia si es necesario cambiar la dosis u otro tratamiento [ver ADVERTENCIAS Y PRECAUCIONES ].
ADVERTENCIAS
Incluido como parte de la PRECAUCIONES sección.
PRECAUCIONES
El embarazo
Las cápsulas de REBETOL de 200 mg y la solución oral pueden causar defectos de nacimiento y la muerte del feto. La terapia con REBETOL no debe iniciarse hasta que se haya obtenido un informe de una prueba de embarazo negativa inmediatamente antes del inicio planificado de la terapia. Los pacientes deben usar al menos dos formas de anticoncepción y hacerse pruebas de embarazo mensuales durante el tratamiento y durante el período de 6 meses después de que se suspenda el tratamiento. Se debe tener sumo cuidado para evitar el embarazo en pacientes mujeres y en las parejas femeninas de pacientes varones. REBETOL 200mg ha demostrado efectos teratogénicos y embriocidas significativos en todas las especies animales en las que se han realizado estudios adecuados. Estos efectos ocurrieron a dosis como tan bajo como una vigésima parte de la dosis humana recomendada de ribavirina. El tratamiento con REBETOL 200 mg no debe iniciarse hasta que se informe de una prueba de embarazo negativa. se ha obtenido inmediatamente antes del inicio planificado de la terapia [ver ADVERTENCIA EN CAJA , CONTRAINDICACIONES , Uso en poblaciones específicas , y INFORMACIÓN DEL PACIENTE ].
Anemia
La principal toxicidad de la ribavirina es la anemia hemolítica, que se observó en aproximadamente el 10 % de los sujetos tratados con REBETOL/INTRON A en los ensayos clínicos. La anemia asociada con las cápsulas de REBETOL ocurre dentro de 1 a 2 semanas del inicio de la terapia. Debido a que la caída inicial de la hemoglobina puede ser significativa, se recomienda obtener la hemoglobina o el hematocrito antes del inicio del tratamiento y en las Semanas 2 y 4 de la terapia, o con mayor frecuencia si está clínicamente indicado. Luego, los pacientes deben ser seguidos según sea clínicamente apropiado [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
Se han informado infartos de miocardio fatales y no fatales en pacientes con anemia causada por REBETOL. Se debe evaluar a los pacientes para detectar enfermedades cardíacas subyacentes antes de iniciar el tratamiento con ribavirina. A los pacientes con enfermedades cardíacas preexistentes se les debe administrar electrocardiogramas antes del tratamiento y se les debe monitorear adecuadamente durante la terapia. Si hay algún deterioro del estado cardiovascular, la terapia debe suspenderse o descontinuarse [ver DOSIFICACIÓN Y ADMINISTRACIÓN ]. Debido a que la enfermedad cardíaca puede empeorar por la anemia inducida por medicamentos, los pacientes con antecedentes de enfermedad cardíaca significativa o inestable no deben usar REBETOL.
pancreatitis
El tratamiento con REBETOL e INTRON A o PegIntron debe suspenderse en pacientes con signos y síntomas de pancreatitis y discontinuarse en pacientes con pancreatitis confirmada.
Trastornos pulmonares
Se han informado síntomas pulmonares, que incluyen disnea, infiltrados pulmonares, neumonitis, hipertensión pulmonar y neumonía, durante el tratamiento con REBETOL con terapia de combinación de interferón alfa; se han producido casos ocasionales de neumonía mortal. Además, se ha informado sarcoidosis o la exacerbación de la sarcoidosis. Si hay evidencia de infiltrados pulmonares o deterioro de la función pulmonar, se debe monitorear de cerca al paciente y, si corresponde, se debe suspender la terapia combinada.
Trastornos Oftalmológicos
La ribavirina se usa en terapia combinada con interferones alfa. Disminución o pérdida de la visión, retinopatía incluyendo edema macular, arteria o vena retiniana, trombosis, hemorragias retinianas y manchas algodonosas, neuritis óptica, papiledema y desprendimiento seroso de retina son inducidos o agravados por el tratamiento con interferones alfa. Todos los pacientes deben recibir un examen ocular al inicio. Los pacientes con trastornos oftalmológicos preexistentes (p. ej., retinopatía diabética o hipertensiva) deben someterse a exámenes oftalmológicos periódicos durante la terapia combinada con interferón alfa. Cualquier paciente que desarrolle síntomas oculares debe recibir un examen ocular rápido y completo. La terapia combinada con interferones alfa debe suspenderse en pacientes que desarrollen trastornos oftalmológicos nuevos o que empeoren.
Pruebas de laboratorio
PegIntron en combinación con ribavirina puede causar disminuciones graves en los recuentos de neutrófilos y plaquetas, y anomalías hematológicas, endocrinas (p. ej., TSH) y hepáticas.
Los pacientes que reciben la terapia combinada de PegIntron/REBETOL deben someterse a pruebas de hematología y bioquímica sanguínea antes del inicio del tratamiento y luego periódicamente. En el ensayo clínico de adultos, se midieron hemogramas completos (incluidos los recuentos de hemoglobina, neutrófilos y plaquetas) y bioquímicos (incluidos AST, ALT, bilirrubina y ácido úrico) durante el período de tratamiento en las semanas 2, 4, 8, 12 y luego a intervalos de 6 semanas, o con más frecuencia si se desarrollaron anomalías. En sujetos pediátricos, se evaluaron los mismos parámetros de laboratorio con una evaluación adicional de hemoglobina en la semana 6 de tratamiento. Los niveles de TSH se midieron cada 12 semanas durante el período de tratamiento. El ARN-VHC debe medirse periódicamente durante el tratamiento [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
Trastornos dentales y periodontales
Se han informado trastornos dentales y periodontales en pacientes que reciben tratamiento combinado con ribavirina e interferón o peginterferón. Además, la boca seca podría tener un efecto dañino en los dientes y las membranas mucosas de la boca durante el tratamiento a largo plazo con la combinación de REBETOL e interferón alfa-2b pegilado o no pegilado. Los pacientes deben cepillarse bien los dientes dos veces al día y someterse a exámenes dentales periódicos. Si se producen vómitos, se les debe recomendar que se enjuaguen bien la boca después.
Administración concomitante de azatioprina
Se ha informado en la literatura que se produce pancitopenia (disminución marcada de glóbulos rojos, neutrófilos y plaquetas) y supresión de la médula ósea dentro de las 3 a 7 semanas posteriores a la administración concomitante de interferón pegilado/ribavirina y azatioprina. En este número limitado de pacientes (n = 8), la mielotoxicidad fue reversible dentro de las 4 a 6 semanas posteriores a la suspensión de la terapia antiviral contra el VHC y la azatioprina concomitante y no volvió a aparecer con la reintroducción de cualquiera de los tratamientos solos. Se debe suspender PegIntron, REBETOL y azatioprina por pancitopenia, y no se debe reintroducir interferón pegilado/ribavirina con azatioprina concomitante [ver INTERACCIONES CON LA DROGAS ].
Impacto en el crecimiento: uso pediátrico
Los datos sobre los efectos de PegIntron y REBETOL en el crecimiento provienen de un estudio abierto en sujetos de 3 a 17 años de edad, en el que se comparan los cambios de peso y estatura con los datos normativos de la población de EE. UU. En general, el aumento de peso y estatura de los sujetos pediátricos tratados con PegIntron y REBETOL 200 mg va a la zaga de lo previsto por los datos normativos de la población durante todo el tratamiento. Se observó una velocidad de crecimiento gravemente inhibida (menos del percentil 3) en el 70 % de los sujetos durante el tratamiento. Después del tratamiento, se produjo un crecimiento de rebote y un aumento de peso en la mayoría de los sujetos. Sin embargo, los datos de seguimiento a largo plazo en sujetos pediátricos indican que PegIntron en terapia combinada con REBETOL puede inducir una inhibición del crecimiento que resulta en una reducción de la estatura adulta en algunos pacientes [ver REACCIONES ADVERSAS ].
De manera similar, se observó un impacto en el crecimiento en sujetos después del tratamiento con REBETOL 200 mg e INTRON A durante un año. En un ensayo de seguimiento a largo plazo de un número limitado de estos sujetos, la terapia de combinación resultó en una reducción de la estatura adulta final en algunos sujetos [ver REACCIONES ADVERSAS ].
Garantías de uso
Según los resultados de los ensayos clínicos, la monoterapia con ribavirina no es eficaz para el tratamiento de la infección crónica por el virus de la hepatitis C; por lo tanto, las cápsulas de 200 mg de REBETOL o la solución oral no deben usarse solos. La seguridad y eficacia de las cápsulas y la solución oral de REBETOL solo se han establecido cuando se usan junto con INTRON A o PegIntron (no con otros interferones) como terapia combinada.
No se ha establecido la seguridad y eficacia de la terapia con REBETOL/INTRON A y PegIntron para el tratamiento de infecciones por VIH, adenovirus, RSV, parainfluenza o influenza. Las cápsulas de REBETOL de 200 mg no deben usarse para estas indicaciones. La ribavirina para inhalación tiene una etiqueta separada, que debe consultarse si se está considerando la terapia de inhalación de ribavirina.
Hay reacciones adversas significativas causadas por la terapia con REBETOL/INTRON A o PegIntron, que incluyen depresión severa e ideación suicida, anemia hemolítica, supresión de la función de la médula ósea, trastornos autoinmunes e infecciosos, disfunción pulmonar, pancreatitis y diabetes. La ideación o los intentos de suicidio ocurrieron con mayor frecuencia entre los pacientes pediátricos, principalmente adolescentes, en comparación con los pacientes adultos (2,4 % frente al 1 %) durante el tratamiento y el seguimiento sin tratamiento. El etiquetado de INTRON A y PegIntron debe revisarse en su totalidad para obtener información de seguridad adicional antes de iniciar el tratamiento combinado.
Información de asesoramiento para pacientes
Aconseje al paciente que lea el etiquetado para el paciente aprobado por la FDA ( Guía de medicamentos ).
Anemia
La experiencia adversa más común que ocurre con las cápsulas de REBETOL es la anemia, que puede ser grave [ver ADVERTENCIAS Y PRECAUCIONES y REACCIONES ADVERSAS ]. Se debe advertir a los pacientes que se requieren evaluaciones de laboratorio antes de comenzar la terapia y periódicamente a partir de entonces [ver DOSIFICACIÓN Y ADMINISTRACIÓN ]. Se recomienda que los pacientes estén bien hidratados, especialmente durante las etapas iniciales del tratamiento.
El embarazo
Se debe informar a los pacientes que las cápsulas de 200 mg de REBETOL y la solución oral pueden causar defectos de nacimiento y la muerte del feto. REBETOL 200 mg no debe ser utilizado por mujeres embarazadas ni por hombres cuyas parejas femeninas estén embarazadas. Se debe tener extremo cuidado para evitar el embarazo en pacientes femeninas y en las parejas femeninas de pacientes masculinos que toman REBETOL. No se debe iniciar REBETOL hasta que se haya obtenido un informe de una prueba de embarazo negativa inmediatamente antes del inicio de la terapia. Los pacientes deben realizar una prueba de embarazo mensualmente durante la terapia y durante los 6 meses posteriores a la terapia.
Las mujeres en edad fértil deben recibir asesoramiento sobre el uso de métodos anticonceptivos eficaces (dos formas fiables) antes de iniciar el tratamiento. Se debe advertir a los pacientes (hombres y mujeres) sobre los riesgos teratogénicos/embriocidas y se les debe instruir para que practiquen métodos anticonceptivos efectivos durante la administración de REBETOL 200 mg y durante los 6 meses posteriores a la terapia. Se debe recomendar a los pacientes (hombres y mujeres) que notifiquen al médico de inmediato en caso de embarazo [ver CONTRAINDICACIONES , ADVERTENCIAS Y PRECAUCIONES , y Uso en poblaciones específicas ].
Si ocurre un embarazo durante el tratamiento o durante los 6 meses posteriores a la terapia, se debe informar a la paciente sobre el riesgo teratogénico de la terapia con REBETOL 200 mg para el feto. Los pacientes, o las parejas de los pacientes, deben informar de inmediato a su médico cualquier embarazo que ocurra durante el tratamiento o dentro de los 6 meses posteriores a la finalización del tratamiento. Los prescriptores deben informar estos casos llamando al 1-800-593-2214.
Riesgos versus beneficios
Se debe informar a los pacientes que reciben cápsulas de REBETOL sobre los beneficios y riesgos asociados con el tratamiento, se les debe indicar su uso apropiado y se les debe derivar al paciente. Guía de medicamentos . Se debe informar a los pacientes que se desconoce el efecto del tratamiento de la infección por hepatitis C en la transmisión y que se deben tomar las precauciones adecuadas para prevenir la transmisión del virus de la hepatitis C.
Se debe informar a los pacientes sobre qué hacer en caso de que olviden una dosis de REBETOL; la dosis olvidada debe tomarse lo antes posible durante el mismo día. Los pacientes no deben duplicar la siguiente dosis. Se debe recomendar a los pacientes que se comuniquen con su proveedor de atención médica si tienen preguntas.
Toxicología no clínica
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
Carcinogénesis
La ribavirina no provocó un aumento en ningún tipo de tumor cuando se administró durante 6 meses en el modelo de ratón transgénico con deficiencia de p53 en dosis de hasta 300 mg/kg (equivalente humano estimado de 25 mg/kg según el ajuste del área de superficie corporal para un adulto de 60 kg). ; aproximadamente 1,9 veces la dosis diaria máxima recomendada en humanos). La ribavirina no fue cancerígena cuando se administró durante 2 años a ratas en dosis de hasta 40 mg/kg (equivalente humano estimado de 5,71 mg/kg basado en el ajuste del área de superficie corporal para un adulto de 60 kg).
mutagénesis
La ribavirina demostró una mayor incidencia de mutación y transformación celular en múltiples ensayos de genotoxicidad. La ribavirina fue activa en el ensayo de transformación celular in vitro Balb/3T3. Se observó actividad mutagénica en el ensayo de linfoma de ratón y en dosis de 20 a 200 mg/kg (equivalente humano estimado de 1,67 a 16,7 mg/kg, basado en el ajuste del área de superficie corporal para un adulto de 60 kg; 0,1 a 1 veces el máximo dosis recomendada de ribavirina para 24 horas en humanos) en un ensayo de micronúcleos de ratón. Un ensayo letal dominante en ratas fue negativo, lo que indica que si ocurrieron mutaciones en ratas, no se transmitieron a través de gametos masculinos.
Deterioro de la fertilidad
La ribavirina demostró efectos embriocidas y teratogénicos significativos a dosis muy por debajo de la dosis humana recomendada en todas las especies animales en las que se han realizado estudios adecuados. Se observaron malformaciones del cráneo, paladar, ojo, mandíbula, extremidades, esqueleto y tracto gastrointestinal. La incidencia y la gravedad de los efectos teratogénicos aumentaron con el aumento de la dosis del fármaco. Se redujo la supervivencia de fetos y crías. En estudios convencionales de embriotoxicidad/teratogenicidad en ratas y conejos, los niveles de dosis sin efecto observados fueron muy inferiores a los del uso clínico propuesto (0,3 mg/kg/día tanto para la rata como para el conejo; aproximadamente 0,06 veces la dosis humana recomendada de 24 horas de ribavirina). No se observó toxicidad materna ni efectos en la descendencia en un estudio de toxicidad peri/postnatal en ratas a las que se administró una dosis oral de hasta 1 mg/kg/día (dosis equivalente humana estimada de 0,17 mg/kg basada en el ajuste del área de superficie corporal para un adulto de 60 kg). ; aproximadamente 0,01 veces la dosis máxima recomendada de ribavirina en humanos durante 24 horas) [ver CONTRAINDICACIONES , y ADVERTENCIAS Y PRECAUCIONES ].
Las mujeres fértiles y las parejas de mujeres fértiles no deben recibir REBETOL a menos que el paciente y su pareja estén usando métodos anticonceptivos efectivos (dos formas confiables). Sobre la base de una vida media de dosis múltiples (t1/2) de ribavirina de 12 días, se debe utilizar un método anticonceptivo eficaz durante 6 meses después de la terapia (p. ej., 15 vidas medias de aclaramiento de ribavirina).
REBETOL debe usarse con precaución en hombres fértiles. En estudios en ratones para evaluar la evolución temporal y la reversibilidad de la degeneración testicular inducida por ribavirina a dosis de 15 a 150 mg/kg/día (equivalente humano estimado de 1,25 a 12,5 mg/kg/día, basado en el ajuste del área de superficie corporal para un adulto de 60 kg; 0,1-0,8 veces la dosis máxima humana de 24 horas de ribavirina) administrada durante 3 o 6 meses, se produjeron anomalías en los espermatozoides. Tras la interrupción del tratamiento, la recuperación esencialmente total de la toxicidad testicular inducida por ribavirina fue evidente en 1 o 2 ciclos de espermatogénesis.
Uso en poblaciones específicas
El embarazo
Embarazo Categoría X
[Ver CONTRAINDICACIONES , ADVERTENCIAS Y PRECAUCIONES , y Toxicología no clínica ].
Tratamiento y Post-tratamiento:
Riesgo potencial para el feto
Se sabe que la ribavirina se acumula en los componentes intracelulares de donde se elimina muy lentamente. No se sabe si la ribavirina contenida en el esperma ejercerá un efecto teratogénico potencial sobre la fertilización de los óvulos. En un estudio en ratas, se concluyó que la letalidad dominante no fue inducida por ribavirina en dosis de hasta 200 mg/kg durante 5 días (dosis equivalentes humanas estimadas de 7,14 a 28,6 mg/kg, con base en el ajuste del área de superficie corporal para un 60 kg adulto; hasta 1,7 veces la dosis máxima recomendada de ribavirina en humanos). Sin embargo, debido a los posibles efectos teratogénicos humanos de la ribavirina, se debe recomendar a los pacientes varones que tomen todas las precauciones necesarias para evitar el riesgo de embarazo para sus parejas femeninas.
Las mujeres en edad fértil no deben recibir REBETOL a menos que estén usando métodos anticonceptivos efectivos (dos formas confiables) durante el período de terapia. Además, se debe utilizar un método anticonceptivo eficaz durante los 6 meses posteriores a la terapia según una vida media (t1/2) de dosis múltiples de ribavirina de 12 días.
Los pacientes masculinos y sus parejas femeninas deben practicar métodos anticonceptivos efectivos (dos formas confiables) durante el tratamiento con REBETOL y durante el período posterior a la terapia de 6 meses (p. ej., 15 semividas para la eliminación de ribavirina del cuerpo).
Se ha establecido un registro de embarazos con ribavirina para monitorear los resultados materno-fetales de los embarazos en pacientes mujeres y parejas mujeres de pacientes varones expuestos a ribavirina durante el tratamiento y durante los 6 meses posteriores a la interrupción del tratamiento. Se alienta a los médicos y pacientes a informar tales casos llamando al 1-800-593-2214.
Madres lactantes
No se sabe si el producto REBETOL se excreta en la leche humana. Debido al potencial de reacciones adversas graves del fármaco en los lactantes, se debe tomar la decisión de suspender la lactancia o retrasar o suspender REBETOL.
Uso pediátrico
No se ha establecido la seguridad y eficacia de REBETOL 200 mg en combinación con PegIntron en pacientes pediátricos menores de 3 años. Para el tratamiento con REBETOL/INTRON A, se debe considerar la evidencia de progresión de la enfermedad, como inflamación y fibrosis hepáticas, así como los factores pronósticos de respuesta, el genotipo del VHC y la carga viral al decidir tratar a un paciente pediátrico. Los beneficios del tratamiento deben sopesarse frente a los resultados de seguridad observados.
Los datos de seguimiento a largo plazo en sujetos pediátricos indican que REBETOL en combinación con PegIntron o con INTRON A puede inducir una inhibición del crecimiento que resulta en una estatura reducida en algunos pacientes [ver ADVERTENCIAS Y PRECAUCIONES y REACCIONES ADVERSAS ].
La ideación o los intentos de suicidio ocurrieron con mayor frecuencia entre pacientes pediátricos, principalmente adolescentes, en comparación con pacientes adultos (2,4 % frente a 1 %) durante el tratamiento y el seguimiento sin tratamiento [ver ADVERTENCIAS Y PRECAUCIONES ]. Al igual que en los pacientes adultos, los pacientes pediátricos experimentaron otros reacciones adversas psiquiátricas (p. ej., depresión, labilidad emocional, somnolencia), anemia y neutropenia [ver ADVERTENCIAS Y PRECAUCIONES ].
Uso geriátrico
Los ensayos clínicos de la terapia con REBETOL/INTRON A o PegIntron no incluyeron un número suficiente de sujetos de 65 años o más para determinar si responden de manera diferente a los sujetos más jóvenes.
Se sabe que REBETOL 200 mg se excreta sustancialmente por vía renal, y el riesgo de reacciones tóxicas a este fármaco puede ser mayor en pacientes con insuficiencia renal. Debido a que los pacientes de edad avanzada a menudo tienen una función renal disminuida, se debe tener cuidado al seleccionar la dosis. Se debe monitorear la función renal y se deben hacer los ajustes de dosis correspondientes. REBETOL 200 mg no debe usarse en pacientes con aclaramiento de creatinina inferior a 50 ml/min [ver CONTRAINDICACIONES ].
En general, las cápsulas de REBETOL de 200 mg deben administrarse con precaución a pacientes de edad avanzada, comenzando con el extremo inferior del rango de dosificación, lo que refleja la mayor frecuencia de disminución de la función hepática y cardíaca, y de enfermedades concomitantes u otra terapia con medicamentos. En ensayos clínicos, los sujetos de edad avanzada tuvieron una mayor frecuencia de anemia (67 %) que los pacientes más jóvenes (28 %) [ver ADVERTENCIAS Y PRECAUCIONES ].
Receptores de trasplantes de órganos
No se ha establecido la seguridad y eficacia de INTRON A y PegIntron solos o en combinación con REBETOL para el tratamiento de la hepatitis C en receptores de trasplante de hígado u otros órganos. En una pequeña (n=16) experiencia de caso no controlada de un solo centro, la insuficiencia renal en receptores de aloinjertos renales que recibieron tratamiento combinado con interferón alfa y ribavirina fue más frecuente de lo esperado según la experiencia previa del centro con receptores de aloinjertos renales que no recibieron tratamiento combinado. La relación de la insuficiencia renal con el rechazo del aloinjerto renal no está clara.
Coinfección por VIH o VHB
No se ha establecido la seguridad y eficacia de PegIntron/REBETOL 200 mg e INTRON A/REBETOL para el tratamiento de pacientes con VHC coinfectados con VIH o VHB.
SOBREDOSIS
Hay experiencia limitada con la sobredosis. Se ha informado la ingestión aguda de hasta 20 g de cápsulas de REBETOL, la ingestión de INTRON A de hasta 120 millones de unidades y dosis subcutáneas de INTRON A de hasta 10 veces las dosis recomendadas. Los efectos principales que se han observado son una mayor incidencia y gravedad de las reacciones adversas relacionadas con el uso terapéutico de INTRON A y REBETOL. Sin embargo, se han informado anomalías de las enzimas hepáticas, insuficiencia renal, hemorragia e infarto de miocardio con la administración de dosis subcutáneas únicas de INTRON A que superan las recomendaciones de dosificación.
No existe un antídoto específico para la sobredosis de 200 mg de INTRON A o REBETOL, y la hemodiálisis y la diálisis peritoneal no son eficaces para el tratamiento de la sobredosis de estos agentes.
CONTRAINDICACIONES
La terapia combinada con REBETOL está contraindicada en:
- mujeres que están embarazadas. REBETOL 200 mg puede causar daño fetal cuando se administra a una mujer embarazada. REBETOL está contraindicado en mujeres que están o pueden quedar embarazadas. Si se usa REBETOL durante el embarazo, o si la paciente queda embarazada mientras toma REBETOL 200 mg, se debe informar a la paciente sobre el peligro potencial para el feto [ver ADVERTENCIAS Y PRECAUCIONES , Uso en poblaciones específicas , y INFORMACIÓN DEL PACIENTE ]
- hombres cuyas parejas femeninas están embarazadas
- pacientes con reacciones de hipersensibilidad conocidas, como síndrome de Stevens-Johnson, necrólisis epidérmica tóxica y eritema multiforme a la ribavirina o a cualquier componente del producto
- pacientes con hepatitis autoinmune
- pacientes con hemoglobinopatías (p. ej., talasemia mayor, anemia de células falciformes)
- pacientes con aclaramiento de creatinina inferior a 50 ml/min. [ver Uso en poblaciones específicas y FARMACOLOGÍA CLÍNICA ]
- La coadministración de REBETOL y didanosina está contraindicada porque aumenta la exposición al metabolito activo de didanosina (didesoxiadenosina 5'-trifosfato). Se han notificado insuficiencia hepática mortal, así como neuropatía periférica, pancreatitis e hiperlactatemia/acidosis láctica sintomática en pacientes que reciben didanosina en combinación con ribavirina [ver INTERACCIONES CON LA DROGAS ].
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
La ribavirina es un agente antiviral [ver Microbiología ].
Farmacocinética
Las propiedades farmacocinéticas de dosis únicas y múltiples en adultos se resumen en la Tabla 11. La ribavirina se absorbió rápida y ampliamente después de la administración oral. Sin embargo, debido al metabolismo de primer paso, la biodisponibilidad absoluta promedió 64% (44%). Hubo una relación lineal entre la dosis y el AUCtf (AUC desde el tiempo cero hasta la última concentración medible) después de dosis únicas de 200 a 1200 mg de ribavirina. La relación entre la dosis y la Cmax fue curvilínea, con tendencia a la asíntota por encima de las dosis únicas de 400 a 600 mg.
Tras múltiples dosis orales, basadas en AUC12hr, se observó una acumulación de ribavirina en plasma de 6 veces. Después de la administración oral de 600 mg dos veces al día, se alcanzó el estado estacionario en aproximadamente 4 semanas, con concentraciones plasmáticas promedio en el estado estacionario de 2200 ng/mL (37%). Tras la interrupción de la dosificación, la semivida media fue de 298 (30 %) horas, lo que probablemente refleja una eliminación lenta de los compartimentos no plasmáticos.
Efecto del antiácido sobre la absorción de ribavirina
La coadministración de cápsulas de REBETOL con un antiácido que contiene magnesio, aluminio y simeticona resultó en una disminución del 14 % en el AUCtf medio de la ribavirina. Se desconoce la relevancia clínica de los resultados de este estudio de dosis única.
Distribución de tejidos
El transporte de ribavirina a compartimentos no plasmáticos se ha estudiado más extensamente en los glóbulos rojos y se ha identificado que se realiza principalmente a través de un transportador de nucleósidos equilibrante de tipo es. Este tipo de transportador está presente en prácticamente todos los tipos de células y puede explicar el extenso volumen de distribución. La ribavirina no se une a las proteínas plasmáticas.
Metabolismo y Excreción
La ribavirina tiene dos vías de metabolismo: (i) una vía de fosforilación reversible en células nucleadas; y (ii) una vía de degradación que implica la desribosilación y la hidrólisis de amida para producir un metabolito de ácido triazol carboxílico. La ribavirina y sus metabolitos triazol carboxamida y ácido triazol carboxílico se excretan por vía renal. Después de la administración oral de 600 mg de 14C-ribavirina, aproximadamente el 61 % y el 12 % de la radiactividad se eliminó en la orina y las heces, respectivamente, en 336 horas. La ribavirina inalterada representó el 17% de la dosis administrada.
Poblaciones Especiales
Disfuncion renal
La farmacocinética de la ribavirina se evaluó después de la administración de una dosis oral única (400 mg) de ribavirina a sujetos no infectados por el VHC con diversos grados de disfunción renal. El valor medio de AUCtf fue tres veces mayor en sujetos con valores de aclaramiento de creatinina entre 10 y 30 ml/min en comparación con los sujetos de control (aclaramiento de creatinina superior a 90 ml/min). En sujetos con valores de depuración de creatinina entre 30 y 60 ml/min, el AUCtf fue dos veces mayor en comparación con los sujetos de control. El aumento del AUCtf parece deberse a la reducción del aclaramiento renal y no renal en estos sujetos. Los ensayos de eficacia de fase 3 incluyeron sujetos con valores de aclaramiento de creatinina superiores a 50 ml/min. La farmacocinética de dosis múltiples de ribavirina no se puede predecir con precisión en pacientes con disfunción renal. La ribavirina no se elimina eficazmente por hemodiálisis.
Los pacientes con aclaramiento de creatinina inferior a 50 ml/min no deben recibir tratamiento con REBETOL [ver CONTRAINDICACIONES ].
disfunción hepática
El efecto de la disfunción hepática se evaluó después de una dosis oral única de ribavirina (600 mg). Los valores medios de AUCtf no fueron significativamente diferentes en sujetos con disfunción hepática leve, moderada o grave (clasificación de Child-Pugh A, B o C) en comparación con los sujetos de control. Sin embargo, los valores medios de Cmax aumentaron con la gravedad de la disfunción hepática y fueron dos veces mayores en sujetos con disfunción hepática grave en comparación con los sujetos de control.
Pacientes de edad avanzada
No se han realizado evaluaciones farmacocinéticas en sujetos de edad avanzada.
Género
No se observaron diferencias farmacocinéticas clínicamente significativas en un ensayo de dosis única de 18 hombres y 18 mujeres.
Pacientes pediátricos
Las propiedades farmacocinéticas de dosis múltiples de REBETOL 200 mg cápsulas e INTRON A en pacientes pediátricos con hepatitis C crónica entre 5 y 16 años de edad se resumen en la Tabla 12. La farmacocinética de REBETOL 200 mg e INTRON A (dosis normalizada) es similar en adultos y temas pediátricos.
No se han determinado las características farmacocinéticas completas de REBETOL solución oral en sujetos pediátricos. Los valores de Cmin de ribavirina fueron similares luego de la administración de REBETOL solución oral o cápsulas de 200 mg de REBETOL durante 48 semanas de terapia en sujetos pediátricos (3 a 16 años de edad).
Se realizó un ensayo clínico en sujetos pediátricos con hepatitis C crónica entre 3 y 17 años de edad en el que se evaluó la farmacocinética de PegIntron y REBETOL (cápsulas y solución oral). En sujetos pediátricos que recibieron una dosis de PegIntron ajustada según el área de superficie corporal de 60 mcg/m²/semana, se predijo que la estimación de la relación logarítmica transformada de la exposición durante el intervalo de dosis sería un 58 % [IC del 90 %: 141 %, 177 %] mayor que observado en adultos que recibieron 1,5 mcg/kg/semana. La farmacocinética de REBETOL (dosis normalizada) en este ensayo fue similar a la informada en un estudio anterior de REBETOL 200 mg en combinación con INTRON A en sujetos pediátricos y adultos.
Efecto de los alimentos sobre la absorción de ribavirina
Tanto el AUCtf como la Cmax aumentaron en un 70 % cuando las cápsulas de REBETOL se administraron con una comida rica en grasas (841 kcal, 53,8 g de grasa, 31,6 g de proteína y 57,4 g de carbohidratos) en un estudio farmacocinético de dosis única [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
Microbiología
Mecanismo de acción
El mecanismo por el cual la ribavirina contribuye a su eficacia antiviral en la clínica no se comprende completamente. La ribavirina tiene actividad antiviral directa en cultivo de tejidos contra muchos virus de ARN. La ribavirina aumenta la frecuencia de mutación en los genomas de varios virus y el trifosfato de ribavirina inhibe la polimerasa del VHC en una reacción bioquímica.
Actividad antiviral en cultivo celular
La actividad antiviral de la ribavirina en el replicón del VHC no se comprende bien y no se ha definido debido a la toxicidad celular de la ribavirina. Se ha observado actividad antiviral directa en cultivos de tejidos de otros virus de ARN. La actividad anti-VHC del interferón se demostró en células que contenían VHC-RNS autorreplicantes (células con replicón de VHC) o infección por VHC.
Resistencia
Los genotipos del VHC muestran una amplia variabilidad en su respuesta a la terapia con interferón humano recombinante pegilado/ribavirina. No se han identificado cambios genéticos asociados con la respuesta variable.
Resistencia cruzada
No se ha informado de resistencia cruzada entre los interferones pegilados/no pegilados y la ribavirina.
Toxicología y Farmacología Animal
Estudios a largo plazo en ratones y ratas [18 a 24 meses; dosis de 20 a 75 y 10 a 40 mg/kg/día, respectivamente (dosis equivalentes humanas estimadas de 1,67 a 6,25 y 1,43 a 5,71 mg/kg/día, respectivamente, según el ajuste del área de superficie corporal para un adulto de 60 kg; aproximadamente 0,1 a 0,4 veces la dosis máxima humana de ribavirina en 24 horas)] han demostrado una relación entre la exposición crónica a la ribavirina y el aumento de la incidencia de lesiones vasculares (hemorragias microscópicas) en ratones. En ratas, se produjo degeneración de la retina en los controles, pero la incidencia aumentó en las ratas tratadas con ribavirina.
En un estudio en el que las crías de rata recibieron dosis posnatales de ribavirina de 10, 25 y 50 mg/kg/día, se produjeron muertes relacionadas con el fármaco a 50 mg/kg (a concentraciones plasmáticas de crías de rata por debajo de las concentraciones plasmáticas humanas a dosis humanas). dosis terapéutica) entre los días 13 y 48 del estudio. Las crías de rata a las que se administró la dosis desde el día 7 hasta el 63 después del nacimiento demostraron una disminución menor relacionada con la dosis en el crecimiento general en todas las dosis, que posteriormente se manifestó como una ligera disminución en el peso corporal, la longitud de la coronilla y la grupa. y longitud del hueso. Estos efectos mostraron evidencia de reversibilidad y no se observaron efectos histopatológicos en el hueso. No se observaron efectos de la ribavirina con respecto al desarrollo neuroconductual o reproductivo.
Estudios clínicos
El estudio clínico 1 evaluó la monoterapia con PegIntron. Ver Etiquetado de PegIntron para obtener información sobre este ensayo.
Terapia combinada de REBETOL/PegIntron
Sujetos Adultos
estudio 2
Un ensayo aleatorizado comparó el tratamiento con dos regímenes de 200 mg de PegIntron/REBETOL [1,5 mcg/kg de PegIntron por vía subcutánea una vez a la semana/800 mg de REBETOL por vía oral al día (en dosis divididas); PegIntron 1,5 mcg/kg por vía subcutánea una vez a la semana durante 4 semanas, luego 0,5 mcg/kg por vía subcutánea una vez a la semana durante 44 semanas/REBETOL 1000 o 1200 mg por vía oral al día (en dosis divididas)] con INTRON A [3 MUI por vía subcutánea tres veces por semana/REBETOL 1000 o 1200 mg por vía oral al día (en dosis divididas)] en 1530 adultos con hepatitis C crónica. Los sujetos sin tratamiento previo con interferón fueron tratados durante 48 semanas y seguidos durante 24 semanas después del tratamiento. Los sujetos elegibles tenían enfermedad hepática compensada, HCV-RNA detectable, ALT elevada e histopatología hepática compatible con hepatitis crónica.
La respuesta al tratamiento se definió como ARN-VHC indetectable 24 semanas después del tratamiento (ver Tabla 13). La tasa de respuesta a la dosis de 1,5 mcg/kg de PegIntron y 800 mg de ribavirina fue mayor que la tasa de respuesta a INTRON A/REBETOL (consulte la Tabla 13). La tasa de respuesta a 1,5→0,5 mcg/kg/REBETOL de PegIntron fue esencialmente la misma como la respuesta a INTRON A/REBETOL (datos no mostrados).
Los sujetos con el genotipo viral 1, independientemente de la carga viral, tuvieron una tasa de respuesta más baja a PegIntron (1,5 mcg/kg)/REBETOL (800 mg) en comparación con sujetos con otros genotipos virales. Los sujetos con ambos factores de mal pronóstico (genotipo 1 y carga viral alta) tuvieron una tasa de respuesta del 30 % (78/256) en comparación con una tasa de respuesta del 29 % (71/247) con la terapia de combinación INTRON A/REBETOL.
Los sujetos con menor peso corporal tendían a tener mayores tasas de reacciones adversas [ver REACCIONES ADVERSAS y mayores tasas de respuesta que los sujetos con mayor peso corporal. Las diferencias en las tasas de respuesta entre los brazos de tratamiento no variaron sustancialmente con el peso corporal.
Las tasas de respuesta al tratamiento con la combinación de PegIntron/REBETOL fueron del 49 % en hombres y del 56 % en mujeres. Las tasas de respuesta fueron más bajas en los sujetos afroamericanos e hispanos y más altas en los asiáticos en comparación con los caucásicos. Aunque los afroamericanos tenían una mayor proporción de factores de mal pronóstico en comparación con los caucásicos, el número de no caucásicos estudiados (11 % del total) fue insuficiente para permitir conclusiones significativas sobre las diferencias en las tasas de respuesta después de ajustar los factores de pronóstico en este ensayo.
Se obtuvieron biopsias de hígado antes y después del tratamiento en el 68% de los sujetos. En comparación con el valor inicial, se observó que aproximadamente dos tercios de los sujetos en todos los grupos de tratamiento tenían una reducción modesta de la inflamación.
Estudio 3
En un gran ensayo basado en la comunidad de los Estados Unidos, 4913 sujetos con hepatitis C crónica fueron aleatorizados para recibir PegIntron 1,5 mcg/kg por vía subcutánea una vez a la semana en combinación con una dosis de 200 mg de REBETOL de 800 a 1400 mg (dosis basada en el peso [WBD]) o 800 mg (flat) por vía oral al día (en dosis divididas) durante 24 o 48 semanas según el genotipo. La respuesta al tratamiento se definió como ARN-VHC indetectable (basado en un ensayo con un límite inferior de detección de 125 UI/mL) a las 24 semanas posteriores al tratamiento.
El tratamiento con 1,5 mcg/kg de PegIntron y 800 a 1400 mg de REBETOL dio como resultado una respuesta virológica sostenida más alta en comparación con PegIntron en combinación con una dosis diaria fija de 800 mg de REBETOL. Los sujetos que pesaban más de 105 kg obtuvieron el mayor beneficio con WBD, aunque también se observó un beneficio modesto en sujetos que pesaban más de 85 a 105 kg (consulte la Tabla 14). El beneficio de WBD en sujetos que pesaban más de 85 kg se observó con los genotipos 1-3 del VHC. No se disponía de datos suficientes para llegar a conclusiones con respecto a otros genotipos. El uso de WBD resultó en una mayor incidencia de anemia [ver REACCIONES ADVERSAS ].
Un total de 1552 sujetos que pesaban más de 65 kg en el Estudio 3 tenían el genotipo 2 o 3 y se aleatorizaron a 24 o 48 semanas de terapia. No se observó ningún beneficio adicional con la mayor duración del tratamiento.
Estudio 4
Un gran ensayo aleatorizado comparó la seguridad y la eficacia del tratamiento durante 48 semanas con dos regímenes de 200 mg de PegIntron/REBETOL [1,5 mcg/kg de PegIntron y 1 mcg/kg por vía subcutánea una vez a la semana, ambos en combinación con 800 a 1400 mg de REBETOL por vía oral al día (en dos dosis)] y Pegasys 180 mcg por vía subcutánea una vez a la semana en combinación con Copegus 1000 a 1200 mg PO al día (en dos dosis divididas) en 3070 adultos sin tratamiento previo con hepatitis C crónica genotipo 1. En este ensayo, la falta de respuesta virológica temprana (indetectable ARN-VHC o una reducción mayor o igual a 2 log10 desde el inicio) por tratamiento La semana 12 fue el criterio para la interrupción del tratamiento. La RVS se definió como ARN-VHC indetectable (ensayo Roche COBAS TaqMan, un límite inferior de cuantificación de 27 UI/mL) a las 24 semanas posteriores al tratamiento (consulte la Tabla 15).
Las tasas generales de RVS fueron similares entre los tres grupos de tratamiento. Independientemente del grupo de tratamiento, las tasas de RVS fueron más bajas en sujetos con factores de mal pronóstico. Sin embargo, los sujetos con factores de mal pronóstico aleatorizados a PegIntron (1,5 mcg/kg)/REBETOL o Pegasys/Copegus lograron tasas de RVS más altas en comparación con sujetos similares aleatorizados a PegIntron 1 mcg/kg/REBETOL. Para la dosis de 1,5 mcg/kg de PegIntron y REBETOL, las tasas de RVS para sujetos con y sin los siguientes factores pronósticos fueron las siguientes: cirrosis (10 % frente a 42 %), niveles normales de ALT (32 % frente a 42 %), carga superior a 600 000 UI/mL (35 % frente a 61 %), mayores de 40 años (38 % frente a 50 %) y raza afroamericana (23 % frente a 44 %). En sujetos con ARN-VHC indetectable en la semana 12 de tratamiento que recibieron PegIntron (1,5 mcg/kg)/200 mg de REBETOL, la tasa de RVS fue del 81 % (328/407).
Estudio 5: terapia combinada de REBETOL/PegIntron en fracasos de tratamientos previos
En un ensayo no comparativo, 2293 sujetos con fibrosis de moderada a grave que fracasaron en el tratamiento previo con una combinación de interferón alfa/ribavirina fueron tratados nuevamente con PegIntron, 1,5 mcg/kg por vía subcutánea, una vez a la semana, en combinación con ribavirina ajustada al peso. Los sujetos elegibles incluyeron no respondedores previos (sujetos que dieron positivo en ARN-VHC al final de un mínimo de 12 semanas de tratamiento) y recaídas previas (sujetos que dieron negativo en VHCARN al final de un mínimo de 12 semanas de tratamiento y posteriormente recayeron después del tratamiento). hacer un seguimiento). Los sujetos que dieron negativo en la semana 12 fueron tratados durante 48 semanas y seguidos durante 24 semanas después del tratamiento. La respuesta al tratamiento se definió como ARN-VHC indetectable a las 24 semanas posteriores al tratamiento (medido mediante una prueba basada en investigaciones, límite de detección de 125 UI/mL). La tasa de respuesta global fue del 22 % (497/2293) (IC del 99 %: 19,5, 23,9). Los sujetos con las siguientes características tenían menos probabilidades de beneficiarse de un nuevo tratamiento: falta de respuesta previa, tratamiento previo con interferón pegilado, fibrosis en puente significativa o cirrosis e infección por genotipo 1.
Las tasas de respuesta virológica sostenida de retratamiento por características iniciales se resumen en la Tabla 16.
El logro de un ARN-VHC indetectable en la semana 12 de tratamiento fue un fuerte predictor de RVS. En este ensayo, 1470 (64 %) sujetos no lograron un ARN-VHC indetectable en la semana 12 de tratamiento y se les ofreció participar en ensayos de tratamiento a largo plazo debido a una respuesta inadecuada al tratamiento. De los 823 (36 %) sujetos en los que el ARN del VHC era indetectable en la semana 12 de tratamiento, los infectados con el genotipo 1 tuvieron una RVS del 48 % (245/507), con un rango de respuestas según las puntuaciones de fibrosis (F4-F2) de 39-55%. Los sujetos infectados con el genotipo 2/3 que tenían ARN-VHC indetectable en la semana 12 de tratamiento tuvieron una RVS general del 70 % (196/281), con un rango de respuestas según las puntuaciones de fibrosis (F4-F2) del 60-83 %. Para todos los genotipos, las puntuaciones más altas de fibrosis se asociaron con una menor probabilidad de alcanzar la RVS.
Sujetos pediátricos
Los sujetos pediátricos de 3 a 17 años de edad sin tratamiento previo con hepatitis C crónica compensada y ARN-VHC detectable fueron tratados con REBETOL 15 mg/kg por día y PegIntron 60 mcg/m² una vez a la semana durante 24 o 48 semanas según el genotipo del VHC y el nivel viral inicial. carga. Todos los sujetos debían ser seguidos durante 24 semanas después del tratamiento. Un total de 107 sujetos recibieron tratamiento, de los cuales el 52 % eran mujeres, el 89 % eran caucásicos y el 67 % estaban infectados con el genotipo 1 del VHC. UI/mL recibieron 48 semanas de terapia mientras que aquellos infectados con Genotipo 2 o Genotipo 3 con HCV-RNA menos de 600 000 UI/mL recibieron 24 semanas de terapia. Los resultados del ensayo se resumen en la Tabla 17.
Tratamiento combinado con REBETOL/INTRON A
Sujetos Adultos
Sujetos no tratados previamente
Adultos con hepatitis C crónica compensada y ARN-VHC detectable (evaluado por un laboratorio central mediante un ensayo de RT-PCR basado en investigaciones) que no habían sido tratados previamente con terapia con interferón alfa se inscribieron en dos ensayos multicéntricos doble ciego (EE. UU. e internacionales) y aleatorizados para recibir cápsulas de REBETOL de 200 mg, 1200 mg/día (1000 mg/día para sujetos que pesen menos o igual a 75 kg) e INTRON A 3 MUI tres veces por semana o INTRON A y placebo durante 24 o 48 semanas seguidas de 24 semanas de seguimiento fuera de la terapia. El ensayo internacional no contenía un brazo de tratamiento con INTRON A de 24 semanas y placebo. El ensayo estadounidense inscribió a 912 sujetos que, al inicio, eran 67 % hombres, 89 % caucásicos con una puntuación media de Knodell HAI (I+II+III) de 7,5 y 72 % genotipo 1. El ensayo internacional, realizado en Europa, Israel , Canadá y Australia, inscribieron a 799 sujetos (65 % hombres, 95 % caucásicos, puntuación media de Knodell de 6,8 y 58 % de genotipo 1).
Los resultados del ensayo se resumen en la Tabla 18.
De los sujetos que no habían alcanzado el ARN del VHC por debajo del límite de detección del ensayo basado en la investigación en la semana 24 del tratamiento con REBETOL/INTRON A, menos del 5 % respondió a 24 semanas adicionales de tratamiento combinado.
Entre los sujetos con el genotipo 1 del VHC tratados con REBETOL/INTRON A que alcanzaron el ARN del VHC por debajo del límite de detección del ensayo basado en la investigación a las 24 semanas, los asignados al azar a 48 semanas de tratamiento tuvieron respuestas virológicas más altas en comparación con los de las 24 semanas. grupo de tratamiento de semana. No se observó un aumento en las tasas de respuesta para sujetos con VHC sin genotipo 1 asignados al azar a la terapia con REBETOL/INTRON A durante 48 semanas en comparación con 24 semanas.
Sujetos de recaída
Los sujetos con hepatitis C crónica compensada y ARN-VHC detectable (evaluado por un laboratorio central mediante un ensayo de RT-PCR basado en investigaciones) que habían recaído después de uno o dos cursos de terapia con interferón (definido como niveles anormales de ALT en suero) se inscribieron en dos ensayos multicéntricos, doble ciego (EE. UU. e internacionales) y aleatorizados para recibir REBETOL 1200 mg/día (1000 mg/día para sujetos que pesan ≤ 75 kg) e INTRON A 3 MUI tres veces por semana o INTRON A y placebo durante 24 semanas seguido de 24 semanas de seguimiento sin tratamiento. El ensayo estadounidense inscribió a 153 sujetos que, al inicio, eran 67 % hombres, 92 % caucásicos con una puntuación media de Knodell HAI (I+II+III) de 6,8 y 58 % genotipo 1. El ensayo internacional, realizado en Europa, Israel , Canadá y Australia, inscribieron a 192 sujetos (64 % hombres, 95 % caucásicos, puntuación media de Knodell de 6,6 y 56 % de genotipo 1). Los resultados del ensayo se resumen en la Tabla 19.
Las respuestas virológicas e histológicas fueron similares entre los sujetos masculinos y femeninos tanto en los ensayos sin tratamiento previo como en los de recaída.
Sujetos pediátricos
Sujetos pediátricos de 3 a 16 años de edad con hepatitis C crónica compensada y ARN-VHC detectable (evaluado por un laboratorio central usando un ensayo de RT-PCR basado en investigaciones) fueron tratados con REBETOL 15 mg/kg por día e INTRON A 3 MUI/ m² tres veces por semana durante 48 semanas seguidas de 24 semanas de seguimiento sin tratamiento. Un total de 118 sujetos recibieron tratamiento, de los cuales el 57 % eran hombres, el 80 % caucásicos y el 78 % de genotipo 1. Los sujetos menores de 5 años recibieron REBETOL solución oral y los de 5 años o más recibieron REBETOL solución oral o cápsulas
Los resultados del ensayo se resumen en la Tabla 20.
Los sujetos con el genotipo viral 1, independientemente de la carga viral, tuvieron una tasa de respuesta más baja a la terapia combinada de INTRON A/REBETOL en comparación con los sujetos con el genotipo no 1, 36 % frente a 81 %. Los sujetos con ambos factores de mal pronóstico (genotipo 1 y carga viral alta) tuvieron una tasa de respuesta del 26 % (13/50).
INFORMACIÓN DEL PACIENTE
No se proporcionó información. por favor refiérase a ADVERTENCIAS Y PRECAUCIONES sección.