Zovirax 200mg, 400mg, 800mg Acyclovir Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Zovirax 800mg y cómo se usa?
Zovirax es un medicamento recetado que se usa para tratar los síntomas del herpes labial (herpes labial) y el herpes genital. Zovirax 800 mg puede usarse solo o con otros medicamentos.
Zovirax 400 mg pertenece a una clase de medicamentos llamados antivirales tópicos.
No se sabe si Zovirax 200 mg es seguro y efectivo en niños menores de 12 años.
¿Cuáles son los posibles efectos secundarios de Zovirax 800mg?
Zovirax 200 mg puede causar efectos secundarios graves que incluyen:
- moretones o sangrado fácil,
- puntos morados o rojos debajo de la piel,
- orinar poco o nada,
- micción dolorosa o difícil,
- hinchazón en los pies o los tobillos,
- sentirse cansado, y
- dificultad para respirar
Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Zovirax incluyen:
- náuseas,
- vómitos,
- Diarrea,
- malestar general,
- dolor de cabeza, y
- dolor en la boca al usar una tableta bucal de aciclovir
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Zovirax. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
DESCRIPCIÓN
INFORMACIÓN RESUMEN DEL PRODUCTO
Substancia de droga
Nombre correcto: aciclovir
Nombre químico: 9-[(2-hidroxietoxi)metil]guanina
Otro nombre: acicloguanosina
Fórmula molecular: C8H11N503
Masa molecular: 225.2 Fórmula estructural:
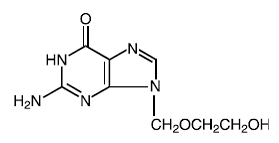
Propiedades fisicoquímicas: El aciclovir es un polvo cristalino blanco con una solubilidad máxima en agua de 1,3 mg/mL a 25°C.
INDICACIONES
Indicaciones y uso clínico
ZOVIRAX® (aciclovir) está indicado para las siguientes condiciones:
- El tratamiento de los episodios iniciales de herpes genital.
- La supresión de recurrencias inusualmente frecuentes de herpes genital (6 o más episodios por año).
- El tratamiento agudo del herpes zoster (culebrilla) y la varicela (varicela).
Los resultados de los estudios clínicos sugieren que algunos pacientes con herpes genital recurrente pueden obtener un beneficio clínico de la administración oral de ZOVIRAX® si se toman al primer signo de un episodio inminente. Los que tienen más probabilidades de beneficiarse son los pacientes que experimentan recurrencias graves y prolongadas; tal terapia intermitente puede ser más apropiada que la terapia de supresión cuando estas recurrencias son infrecuentes.
El tratamiento temprano del herpes zóster agudo (culebrilla) en personas inmunocompetentes con ZOVIRAX® oral resultó en una disminución de la excreción viral; disminución del tiempo de curación; menor difusión; y alivio del dolor agudo.
El tratamiento de la varicela (chickenpox) en pacientes inmunocompetentes con ZOVIRAX® oral redujo el número total de lesiones, aceleró la progresión de las lesiones a las etapas de formación de costras y curación, y disminuyó el número de lesiones hipopigmentadas residuales. Además, ZOVIRAX® disminuyó la fiebre y los síntomas constitucionales asociados con la varicela.
No se ha establecido el uso profiláctico de aciclovir en la varicela.
Geriatría (≥ 65 Años): El uso en la población geriátrica puede estar asociado con diferencias en la seguridad debido a los cambios en la función renal relacionados con la edad y se puede encontrar una breve discusión en las secciones correspondientes (ver ADVERTENCIAS Y PRECAUCIONES ).
Pediatría ( No hay datos disponibles.
DOSIFICACIÓN Y ADMINISTRACIÓN
Consideraciones de dosificación
- La dosis de ZOVIRAX® (aciclovir) debe reducirse en pacientes con insuficiencia renal.
- La terapia debe iniciarse lo antes posible después de un diagnóstico de varicela o herpes zoster, o ante el primer signo o síntoma de un brote de herpes genital.
- La dosis recomendada y la duración del uso dependen de la indicación.
Dosis recomendada y ajuste de dosis
Tratamiento de la infección inicial de herpes genital: 200 mg (una tableta de 200 mg o una cucharadita de suspensión [5 ml]) cada 4 horas, 5 veces al día para un total de 1 g al día durante 10 días. El tratamiento debe iniciarse lo antes posible tras la aparición de los signos y síntomas.
Terapia supresiva para el herpes genital recurrente
La dosis inicial recomendada es de 200 mg (una tableta de 200 mg o una cucharadita de suspensión [5 ml]) tres veces al día. Esto se puede aumentar si ocurre un avance hasta una dosis de una tableta de 200 mg o una cucharadita [5 ml] de suspensión, cinco veces al día. Si es necesario, se puede considerar una dosis de 400 mg (dos tabletas de 200 mg o dos cucharaditas de suspensión [10 ml]) administrada dos veces al día. Se recomienda una reevaluación periódica de la necesidad de terapia.
La administración de ZOVIRAX® para terapia intermitente es de 200 mg (una tableta de 200 mg o una cucharadita [5 ml] de suspensión) cada 4 horas, 5 veces al día durante 5 días. La terapia debe iniciarse ante el primer signo o síntoma (pródromo) de recurrencia.
Tratamiento del herpes zoster
800 mg de ZOVIRAX® oral, cada 4 horas, 5 veces al día durante 7 a 10 días. El tratamiento debe iniciarse dentro de las 72 horas de la aparición de las lesiones. En los ensayos clínicos, el mayor beneficio se produjo cuando el tratamiento se inició dentro de las 48 horas siguientes a la aparición de las lesiones.
Tratamiento de la varicela
20 mg/kg (sin exceder los 800 mg) por vía oral, 4 veces al día durante 5 días. La terapia debe iniciarse dentro de las 24 horas posteriores a la aparición de la erupción.
Pacientes con Insuficiencia Renal Aguda o Crónica
Se recomienda precaución cuando se administre aciclovir a pacientes con insuficiencia renal. Se debe mantener una hidratación adecuada.
Se han completado estudios farmacocinéticos exhaustivos después de infusiones intravenosas de aciclovir en pacientes con insuficiencia renal.
Con base en estos estudios, se recomiendan ajustes de dosis en la Tabla 5 para las indicaciones de herpes genital y herpes zoster.
Hemodiálisis
Para los pacientes que requieren hemodiálisis, la vida media plasmática media del aciclovir durante la hemodiálisis es de aproximadamente 5 horas. Esto da como resultado una disminución del 60 % en las concentraciones plasmáticas después de un período de diálisis de seis horas. Por lo tanto, el horario de dosificación del paciente debe ajustarse para que se administre una dosis adicional después de cada diálisis.
Diálisis peritoneal
No parece ser necesaria una dosis de suplemento después del ajuste del intervalo de dosificación.
Dosis olvidada
Si se olvida una dosis de ZOVIRAX®, se debe recomendar al paciente que la tome tan pronto como se acuerde y luego continúe con la siguiente dosis en el intervalo de tiempo adecuado.
CÓMO SUMINISTRADO
Almacenamiento y estabilidad
Las tabletas de ZOVIRAX® deben almacenarse a temperatura ambiente controlada (15 a 25°C) en un lugar seco y protegido de la luz.
ZOVIRAX® Suspensión debe almacenarse a temperatura ambiente controlada (15 a 25 °C).
Formas Farmacéuticas, Composición Y Envases
Suspensión: Cada cucharadita (5 ml) de suspensión ZOVIRAX® contiene 200 mg de aciclovir y los ingredientes no medicinales sabor a plátano, celulosa, glicerina, metilparabeno, propilparabeno, sorbitol, vainillina y agua.
Cada tableta ZOVIRAX® 200 contiene 200 mg de aciclovir y los ingredientes no medicinales celulosa, indigotina, lactosa, estearato de magnesio, povidona y glicolato de almidón de sodio.
ZOVIRAX® Suspensión está disponible en frascos de 125 mL* y 475 mL. Cada cucharadita (5 ml) de suspensión blanquecina con sabor a plátano contiene 200 mg de aciclovir.
*Botella de 125 ml no disponible en Canadá
ZOVIRAX® 200 Comprimidos están disponibles en frascos de 100 comprimidos. Cada tableta comprimida azul, en forma de escudo y con bordes biselados contiene 200 mg de aciclovir y está impresa con "ZOVIRAX" en un lado y un triángulo en el reverso.
GlaxoSmithKline Inc., 7333 Mississauga Road, Mississauga, Ontario, L5N 6L4 1-800-387-7374. Revisado: 10 de noviembre de 2014
EFECTOS SECUNDARIOS
Resumen de reacciones adversas a medicamentos
Las reacciones adversas más frecuentes asociadas al uso de ZOVIRAX® (aciclovir) son dolor de cabeza y náuseas.
También se han informado efectos secundarios neurológicos en raras ocasiones. Los pacientes de edad avanzada y los pacientes con antecedentes de insuficiencia renal tienen un mayor riesgo de desarrollar estos efectos. En los casos notificados, estas reacciones fueron generalmente reversibles al suspender el tratamiento (ver ADVERTENCIAS Y PRECAUCIONES y REACCIONES ADVERSAS , Reacciones adversas a medicamentos posteriores a la comercialización ).
Reacciones adversas a medicamentos en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy específicas, es posible que las tasas de reacciones adversas observadas en los ensayos clínicos no reflejen las tasas observadas en la práctica y no deben compararse con las tasas de los ensayos clínicos de otro fármaco. La información sobre las reacciones adversas a los medicamentos de los ensayos clínicos es útil para identificar los eventos adversos relacionados con los medicamentos y para aproximar las tasas.
Tratamiento del herpes simple
Administración a corto plazo (5-10 días): Las reacciones adversas más frecuentes informadas durante los ensayos clínicos del tratamiento del herpes genital con ZOVIRAX® oral en 298 pacientes se enumeran en la Tabla 1.
Supresión del herpes simple
Administración a largo plazo: Los eventos adversos más frecuentes informados en un ensayo clínico para la prevención de recurrencias con la administración continua de 400 mg (dos cápsulas de 200 mg) 2 veces al día se enumeran en la Tabla 2.
La evidencia hasta ahora de los ensayos clínicos sugiere que es poco probable que la gravedad y la frecuencia de los eventos adversos requieran la interrupción de la terapia.
Infección de herpes
Las reacciones adversas más frecuentes informadas durante tres ensayos clínicos de tratamiento del herpes zoster (culebrilla) con 800 mg de ZOVIRAX® oral 5 veces al día durante 7 o 10 días o placebo se enumeran en la Tabla 3.
Varicela
Los eventos adversos más frecuentes informados durante tres ensayos clínicos de tratamiento de la varicela con ZOVIRAX® oral o placebo se enumeran en la Tabla 4.
Reacciones adversas a medicamentos menos frecuentes en ensayos clínicos (
Otras reacciones adversas informadas en menos del 1 % de los pacientes que recibieron ZOVIRAX® en cualquier ensayo clínico incluyeron: dolor abdominal, anorexia, estreñimiento, mareos, edema, fatiga, flatulencia, adenopatía inguinal, insomnio, dolor en las piernas, sabor a medicamento, erupción cutánea, dolor garganta, movimiento espasmódico de las manos y urticaria.
Hallazgos hematológicos y de química clínica anormales
No se han observado cambios clínicamente significativos en los valores de laboratorio en los ensayos clínicos para el tratamiento de la varicela y el zoster, y para el tratamiento y supresión del herpes genital con ZOVIRAX®.
Reacciones adversas a medicamentos posteriores a la comercialización
Los siguientes eventos se informaron voluntariamente durante el uso posterior a la comercialización de ZOVIRAX® en la práctica clínica. Estos eventos se eligieron para su inclusión debido a su gravedad, frecuencia de notificación, conexión causal potencial con ZOVIRAX® o una combinación de estos factores. Los eventos adversos posteriores a la comercialización se informan espontáneamente de una población de tamaño desconocido, por lo que no se pueden hacer estimaciones de frecuencia.
General: Fiebre, cefalea, dolor y edema periférico.
Nervioso: Se han notificado mareos, parestesia, agitación, confusión, temblor, ataxia, disartria, alucinaciones, síntomas psicóticos, convulsiones, somnolencia, encefalopatía y coma. Estos eventos generalmente son reversibles y generalmente se informan en pacientes con insuficiencia renal o con otros factores predisponentes (ver ADVERTENCIAS Y PRECAUCIONES ). Estos síntomas pueden ser marcados, particularmente en adultos mayores.
Digestivo: Diarrea, malestar gastrointestinal y náuseas.
Hematológico y Linfático: Anemia, leucopenia, linfadenopatía y trombocitopenia.
Hipersensibilidad y Piel: Alopecia, eritema multiforme, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, erupciones que incluyen fotosensibilidad, prurito, urticaria, disnea, angioedema y anafilaxia.
Tracto hepatobiliar y páncreas: Informes de hiperbilirrubinemia reversible y enzimas hepáticas elevadas. Hepatitis e ictericia.
Musculoesquelético: Mialgia.
Sentidos especiales: Anomalías visuales.
urogenitales: Niveles elevados de creatinina en sangre y nitrógeno ureico en sangre (BUN). Se han notificado casos de insuficiencia renal aguda, dolor renal y hematuria. El dolor renal puede estar asociado con insuficiencia renal (ver ADVERTENCIAS Y PRECAUCIONES ).
INTERACCIONES CON LA DROGAS
Interacciones fármaco-fármaco
No se han identificado interacciones clínicamente significativas.
El aciclovir se elimina principalmente sin cambios en la orina a través de la secreción tubular renal activa. Cualquier fármaco administrado simultáneamente que compita con este mecanismo puede aumentar las concentraciones plasmáticas de aciclovir. El probenecid y la cimetidina aumentan el área bajo la curva (AUC) del aciclovir por este mecanismo y reducen el aclaramiento renal del aciclovir. De manera similar, se han demostrado aumentos en las AUC plasmáticas de aciclovir y del metabolito inactivo de micofenolato mofetilo, un agente inmunosupresor utilizado en pacientes trasplantados cuando se administran conjuntamente los fármacos. Sin embargo, no es necesario ajustar la dosis debido al amplio índice terapéutico del aciclovir.
Interacciones entre medicamentos y alimentos
No se conoce ninguna interacción con los alimentos (ver Acción y farmacología clínica , Farmacocinética ).
Interacciones de drogas y hierbas
No se han establecido interacciones con productos herbales.
Interacciones entre fármacos y pruebas de laboratorio
No se han establecido interacciones con pruebas de laboratorio.
ADVERTENCIAS
Las cápsulas, comprimidos y suspensión de ZOVIRAX (aciclovir) están destinados únicamente a la ingestión oral. Se ha observado insuficiencia renal, en algunos casos con resultado de muerte, con la terapia con aciclovir (ver REACCIONES ADVERSAS: Observadas Durante la Práctica Clínica y SOBREDOSIFICACIÓN ). Púrpura trombocitopénica trombótica/síndrome urémico hemolítico (PTT/SUH), que ha provocado la muerte, ha ocurrido en pacientes inmunocomprometidos que reciben terapia con aciclovir.
PRECAUCIONES
Se recomienda ajustar la dosis cuando se administra ZOVIRAX (aciclovir) a pacientes con insuficiencia renal (ver DOSIFICACIÓN Y ADMINISTRACIÓN ). También se debe tener precaución al administrar ZOVIRAX (aciclovir) a pacientes que reciben agentes potencialmente nefrotóxicos, ya que esto puede aumentar el riesgo de disfunción renal y/o el riesgo de síntomas reversibles del sistema nervioso central, como los que se han informado en pacientes tratados con aciclovir intravenoso. . Se debe mantener una hidratación adecuada.
Infección de herpes: No hay datos sobre el tratamiento iniciado más de 72 horas después de la aparición de la erupción por zoster. Se debe recomendar a los pacientes que inicien el tratamiento lo antes posible después de un diagnóstico de herpes zóster.
Infecciones por herpes genital: Se debe informar a los pacientes que ZOVIRAX (aciclovir) no es una cura para el herpes genital. No hay datos que evalúen si ZOVIRAX (aciclovir) evitará la transmisión de la infección a otras personas. Debido a que el herpes genital es una enfermedad de transmisión sexual, los pacientes deben evitar el contacto con las lesiones o las relaciones sexuales cuando las lesiones y/o los síntomas están presentes para evitar infectar a sus parejas. El herpes genital también puede transmitirse en ausencia de síntomas a través de la diseminación viral asintomática. Si está indicado el tratamiento médico de una recurrencia del herpes genital, se debe recomendar a los pacientes que inicien el tratamiento ante el primer signo o síntoma de un episodio.
Varicela: La varicela en niños por lo demás sanos suele ser una enfermedad autolimitada de gravedad leve a moderada. Los adolescentes y adultos tienden a tener una enfermedad más grave. El tratamiento se inició dentro de las 24 horas posteriores a la erupción cutánea típica de la varicela en los estudios controlados, y no hay información sobre los efectos del tratamiento iniciado más tarde en el curso de la enfermedad.
Carcinogénesis, mutagénesis, deterioro de la fertilidad
Los datos que se presentan a continuación incluyen referencias a las concentraciones máximas de aciclovir en plasma en estado estacionario observadas en humanos tratados con 800 mg administrados por vía oral 5 veces al día (dosificación adecuada para el tratamiento del herpes zoster) o 200 mg administrados por vía oral 5 veces al día (dosis adecuada para el tratamiento de herpes genital). Las concentraciones plasmáticas del fármaco en estudios con animales se expresan como múltiplos de la exposición humana al aciclovir en los programas de dosificación más alta y más baja (ver FARMACOLOGÍA CLÍNICA: Farmacocinética ).
El aciclovir se probó en bioensayos de por vida en ratas y ratones en dosis únicas diarias de hasta 450 mg/kg administradas por sonda. No hubo diferencias estadísticamente significativas en la incidencia de tumores entre los animales tratados y los de control, ni el aciclovir acortó la latencia de los tumores. Las concentraciones plasmáticas máximas fueron de 3 a 6 veces los niveles humanos en el bioensayo en ratones y de 1 a 2 veces los niveles humanos en el bioensayo en ratas.
El aciclovir se probó en 16 ensayos de toxicidad genética in vitro e in vivo. El aciclovir fue positivo en 5 de los ensayos.
El aciclovir no afectó la fertilidad o la reproducción en ratones (450 mg/kg/día, po) o en ratas (25 mg/kg/día, sc). En el estudio con ratones, los niveles plasmáticos fueron de 9 a 18 veces los niveles humanos, mientras que en el estudio con ratas, fueron de 8 a 15 veces los niveles humanos. A dosis más altas (50 mg/kg/día, sc) en ratas y conejos (11 a 22 y 16 a 31 veces los niveles humanos, respectivamente) disminuyó la eficacia de implantación, pero no el tamaño de la camada. En un estudio perinatal y posnatal en ratas con 50 mg/kg/día, sc, hubo una disminución estadísticamente significativa en el número medio de cuerpos lúteos, sitios de implantación totales y fetos vivos del grupo.
No se observaron anomalías testiculares en perros que recibieron 50 mg/kg/día, IV durante 1 mes (21 a 41 veces los niveles humanos) o en perros que recibieron 60 mg/kg/día por vía oral durante 1 año (6 a 12 veces los niveles humanos). Se observaron atrofia testicular y aspermatogénesis en ratas y perros a niveles de dosis más altos.
El embarazo
Efectos teratogénicos: Embarazo Categoría B. Acyc lovir administrado durante la organogénesis no fue teratogénico en el ratón (450 mg/kg/día, po), conejo (50 mg/kg/día, sc e IV) o rata (50 mg/kg/día, Carolina del Sur). Estas exposiciones resultaron en niveles plasmáticos de 9 y 18, 16 y 106 y 11 y 22 veces, respectivamente, los niveles humanos.
No existen estudios adecuados y bien controlados en mujeres embarazadas. Un registro epidemiológico prospectivo del uso de aciclovir durante el embarazo se estableció en 1984 y se completó en abril de 1999. Hubo 749 embarazos seguidos en mujeres expuestas al aciclovir sistémico durante el primer trimestre del embarazo, lo que resultó en 756 resultados. La tasa de ocurrencia de defectos de nacimiento se aproxima a la que se encuentra en la población general. Sin embargo, el pequeño tamaño del registro es insuficiente para evaluar el riesgo de defectos menos comunes o para permitir conclusiones confiables o definitivas sobre la seguridad del aciclovir en mujeres embarazadas y sus fetos en desarrollo. El aciclovir debe usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Madres lactantes
Se documentaron concentraciones de aciclovir en la leche materna en 2 mujeres luego de la administración oral de ZOVIRAX (aciclovir) y oscilaron entre 0,6 y 4,1 veces los niveles plasmáticos correspondientes. Estas concentraciones podrían exponer al lactante a una dosis de aciclovir de hasta 0,3 mg/kg/día. ZOVIRAX (aciclovir) debe administrarse a una madre lactante con precaución y solo cuando esté indicado.
Uso pediátrico
No se ha establecido la seguridad y eficacia de las formulaciones orales de aciclovir en pacientes pediátricos menores de 2 años.
Uso geriátrico
De 376 sujetos que recibieron ZOVIRAX (aciclovir) en un estudio clínico del tratamiento del herpes zoster en sujetos inmunocompetentes de 50 años de edad, 244 tenían 65 años o más, mientras que 111 tenían 75 años o más. No se informaron diferencias generales en la efectividad para el tiempo hasta el cese de la formación de nuevas lesiones o el tiempo hasta la curación entre sujetos geriátricos y sujetos adultos más jóvenes. La duración del dolor después de la curación fue mayor en pacientes de 65 años o más. Las náuseas, los vómitos y los mareos se informaron con mayor frecuencia en sujetos de edad avanzada. Los pacientes de edad avanzada tienen más probabilidades de tener una función renal reducida y requieren una reducción de la dosis. Los pacientes de edad avanzada también tienen más probabilidades de sufrir efectos adversos renales o del SNC. Con respecto a los eventos adversos del SNC observados durante la práctica clínica, la somnolencia, las alucinaciones, la confusión y el coma se informaron con mayor frecuencia en pacientes de edad avanzada (ver FARMACOLOGÍA CLÍNICA, REACCIONES ADVERSAS: Observadas durante la práctica clínica, y DOSIFICACIÓN Y ADMINISTRACIÓN ).
SOBREDOSIS
Para el manejo de una sospecha de sobredosis de drogas, comuníquese con su Centro de Control de Toxicología regional.
Se puede administrar carbón activado para ayudar a eliminar el fármaco no absorbido. Se recomiendan medidas generales de apoyo.
El aciclovir se absorbe solo parcialmente en el tracto gastrointestinal. Los pacientes han ingerido hasta 20 g de aciclovir en una sola ocasión, sin efectos adversos inesperados. En estudios clínicos, la concentración plasmática más alta observada en un solo paciente con estas dosis fue de 10,0 μg/mL. Las sobredosis accidentales repetidas de aciclovir oral durante varios días se han asociado con efectos gastrointestinales (como náuseas y vómitos) y efectos neurológicos (dolor de cabeza y confusión).
Las dosis intravenosas administradas a humanos han sido tan altas como 1200 mg/m² (28 mg/kg) 3 veces al día durante un máximo de 2 semanas. Las concentraciones plasmáticas máximas han alcanzado 80 μg/mL. La sobredosis de aciclovir intravenoso ha resultado en elevaciones de la creatinina sérica, nitrógeno ureico en sangre y la subsiguiente insuficiencia renal. Se han descrito efectos neurológicos que incluyen confusión, alucinaciones, agitación, convulsiones y coma en asociación con la sobredosis intravenosa.
Los pacientes deben ser observados de cerca en busca de signos de toxicidad. La hemodiálisis mejora significativamente la eliminación de aciclovir de la sangre y, por lo tanto, puede considerarse una opción de tratamiento en caso de sobredosis sintomática. Puede ocurrir precipitación de aciclovir en los túbulos renales si se excede la solubilidad (2,5 mg/mL) en el líquido intratubular. En caso de insuficiencia renal y anuria, el paciente puede beneficiarse de la hemodiálisis hasta que se restablezca la función renal (ver DOSIFICACIÓN Y ADMINISTRACIÓN ).
CONTRAINDICACIONES
ZOVIRAX® (aciclovir) está contraindicado para pacientes que desarrollan hipersensibilidad o que son hipersensibles al aciclovir, valaciclovir o cualquier otro componente de las formulaciones de ZOVIRAX®. Para obtener una lista completa, consulte Formas de dosificación , Composición y Envasado sección de la monografía del producto .
FARMACOLOGÍA CLÍNICA
Acción y farmacología clínica
Mecanismo de acción
ZOVIRAX® (aciclovir), un análogo de nucleósido de purina acíclico sintético, es un sustrato con un alto grado de especificidad para la timidina quinasa especificada por el herpes simple y la varicela-zóster. El aciclovir es un sustrato deficiente para la timidina quinasa específica de la célula huésped. La timidina quinasa especificada por el herpes simple y la varicela zóster transforma el aciclovir en su monofosfato, que luego se transforma mediante una serie de enzimas celulares en aciclovir difosfato y aciclovir trifosfato. El trifosfato de aciclovir es tanto un inhibidor como un sustrato para la polimerasa de ADN especificada por herpesvirus. Aunque la α-ADN polimerasa celular en las células infectadas también puede ser inhibida por el trifosfato de aciclovir, esto ocurre solo a concentraciones de trifosfato de aciclovir que son más altas que las que inhiben la ADN polimerasa específica del herpesvirus. El aciclovir se convierte selectivamente a su forma activa en las células infectadas con herpesvirus y, por lo tanto, estas células lo captan preferentemente. El aciclovir ha demostrado un potencial tóxico mucho más bajo in vitro para las células normales no infectadas porque: 1) se absorbe menos; 2) menos se convierte a la forma activa; y 3) la α-ADN polimerasa celular tiene una menor sensibilidad a la acción de la forma activa del fármaco. Una combinación de la especificidad de la timidina quinasa, la inhibición de la ADN polimerasa y la terminación prematura de la síntesis de ADN da como resultado la inhibición de la replicación del virus del herpes. No se ha demostrado ningún efecto sobre el virus latente que no se replica. La inhibición del virus reduce el período de diseminación viral, limita el grado de propagación y el nivel de patología y, por lo tanto, facilita la curación. Durante la supresión no hay evidencia de que el aciclovir prevenga la migración neural del virus. Aborta episodios de herpes recurrente debido a la inhibición de la replicación viral después de la reactivación.
Farmacocinética
La farmacocinética de aciclovir después de la administración oral se evaluó en 6 estudios clínicos en los que participaron 110 pacientes adultos.
Absorción
En un estudio de 35 pacientes inmunocomprometidos con infección por herpes simple o varicela zóster que recibieron ZOVIRAX® Cápsulas en dosis de 200 a 1000 mg cada 4 horas, 6 veces al día durante 5 días, se estimó que la biodisponibilidad era del 15 al 20 %. En este estudio, los niveles plasmáticos de estado estacionario se alcanzaron el segundo día de dosificación. Las concentraciones medias máximas y mínimas en estado estacionario después de la última dosis de 200 mg fueron de 0,49 μg/mL (0,47 a 0,54 μg/mL) y 0,31 μg/mL (0,18 a 0,41 μg/mL), respectivamente, y después de la última dosis de 800 mg fueron 2,8 μg/mL (2,3 a 3,1 μg/mL) y 1,8 μg/mL (1,3 a 2,5 μg/mL). En otro estudio, 20 pacientes inmunocompetentes con infecciones genitales recurrentes por herpes simple que recibieron ZOVIRAX® Cápsulas en dosis de 800 mg cada 6 horas, 4 veces al día durante 5 días, las concentraciones medias máximas y mínimas en el estado estacionario fueron de 1,4 μg/ml (0,66 a 1,8 μg/mL) y 0,55 μg/mL (0,14 a 1,1 μg/mL).
En un estudio cruzado de dosis múltiples en el que 23 voluntarios recibieron ZOVIRAX® en una cápsula de 200 mg, una tableta de 400 mg y una tableta de 800 mg 6 veces al día, la absorción disminuyó con el aumento de la dosis y las biodisponibilidades estimadas de aciclovir fueron del 20, 15 y 10 %. , respectivamente. Se cree que la disminución de la biodisponibilidad es una función de la dosis y no de la forma de dosificación. Se demostró que el aciclovir no es proporcional a la dosis en el rango de dosificación de 200 a 800 mg. En este estudio, las concentraciones máxima y mínima de aciclovir en estado estacionario fueron de 0,83 y 0,46 μg/mL, 1,21 y 0,63 μg/mL y 1,61 y 0,83 μg/mL para los regímenes de dosificación de 200, 400 y 800 mg, respectivamente.
En otro estudio en 6 voluntarios, la influencia de los alimentos en la absorción de aciclovir no fue evidente.
Un estudio de biodisponibilidad de dosis oral única en 23 voluntarios normales mostró que las cápsulas de 200 mg de ZOVIRAX® son bioequivalentes a 200 mg de aciclovir en solución acuosa. En un estudio separado en 20 voluntarios, se demostró que la suspensión ZOVIRAX® es bioequivalente a las cápsulas ZOVIRAX®. En un estudio diferente de biodisponibilidad/bioequivalencia de dosis única en 24 voluntarios, se demostró que una tableta de 800 mg de ZOVIRAX® era bioequivalente a cuatro cápsulas de 200 mg de ZOVIRAX®.
Distribución
La unión a proteínas plasmáticas es relativamente baja (9 a 33%) y no se prevén interacciones farmacológicas que impliquen el desplazamiento del sitio de unión.
Eliminación
Después de la administración oral, la vida media plasmática media de aciclovir en voluntarios y pacientes con función renal normal osciló entre 2,5 y 3,3 horas. La excreción renal media del fármaco inalterado representa el 14,4 % (8,6 a 19,8 %) de la dosis administrada por vía oral. El único metabolito urinario (identificado por cromatografía líquida de alta resolución) es la 9-[(carboximetoxi)metil]guanina.
Poblaciones y condiciones especiales
Pediatría
En general, la farmacocinética del aciclovir en niños es similar a la de los adultos. La vida media media después de dosis orales de 300 y 600 mg/m², en niños de 7 meses a 7 años, fue de 2,6 horas (rango de 1,59 a 3,74 horas).
El aciclovir administrado por vía oral en niños menores de 2 años aún no se ha estudiado completamente.
Geriatría
En los ancianos, el aclaramiento corporal total cae con la edad, asociado con disminuciones en el aclaramiento de creatinina, aunque hay pocos cambios en la vida media plasmática terminal. Puede ser necesaria una reducción de la dosis en pacientes geriátricos con función renal reducida (ver DOSIFICACIÓN Y ADMINISTRACIÓN ).
Insuficiencia renal
La vida media y la depuración corporal total del aciclovir dependen de la función renal.
Se recomienda un ajuste de dosis para pacientes con función renal reducida (ver DOSIFICACIÓN Y ADMINISTRACIÓN ).
Ensayos clínicos
Herpes genital inicial
Los estudios doble ciego controlados con placebo han demostrado que ZOVIRAX® administrado por vía oral reduce significativamente la duración de la infección aguda y la duración de la cicatrización de la lesión. La duración del dolor y la formación de nuevas lesiones disminuyó en algunos grupos de pacientes.
Herpes genital recurrente
En un estudio de pacientes que recibieron ZOVIRAX® 400 mg dos veces al día durante 3 años, el 45, 52 y 63 % de los pacientes permanecieron libres de recurrencias en el primer, segundo y tercer año, respectivamente. Los análisis en serie de las tasas de recurrencia de 3 meses para los pacientes mostraron que del 71 al 87 % estaban libres de recurrencia en cada trimestre.
Infecciones por herpes zoster
En un estudio doble ciego controlado con placebo de pacientes inmunocompetentes con infección cutánea por zoster localizada, ZOVIRAX® (800 mg 5 veces al día durante 10 días) acortó los tiempos hasta la formación de costras, la cicatrización y el cese completo del dolor de la lesión, y redujo la duración de la infección viral. desprendimiento y la duración de la formación de nuevas lesiones.
En un estudio doble ciego controlado con placebo similar, ZOVIRAX® (800 mg 5 veces al día durante 7 días) acortó los tiempos para completar la formación de costras, la cicatrización y el cese del dolor de la lesión, y redujo la duración de la formación de nuevas lesiones.
El tratamiento se inició dentro de las 72 horas del inicio de la erupción y fue más efectivo si se inició dentro de las primeras 48 horas. Los adultos mayores de 50 años mostraron un mayor beneficio.
Varicela
Se realizaron tres ensayos aleatorios, doble ciego, controlados con placebo en 993 pacientes pediátricos de 2 a 18 años con varicela. Todos los pacientes fueron tratados dentro de las 24 horas posteriores al inicio de la erupción. En dos ensayos, ZOVIRAX® se administró a 20 mg/kg cuatro veces al día (hasta 3200 mg por día) durante 5 días. En el tercer ensayo, se administraron dosis de 10, 15 o 20 mg/kg cuatro veces al día durante 5 a 7 días. El tratamiento con ZOVIRAX® acortó el tiempo de curación al 50 %, redujo el número máximo de lesiones, redujo la mediana del número de vesículas, disminuyó la mediana del número de lesiones residuales el día 28 y disminuyó la proporción de pacientes con fiebre, anorexia y letargo al día 2. El tratamiento con ZOVIRAX® no afectó las respuestas inmunitarias humorales o celulares específicas del virus varicela zoster al mes o al año después del tratamiento.
Farmacología detallada
Ver Acción y Farmacología Clínica.
Virología
La relación cuantitativa entre la susceptibilidad in vitro del virus del herpes simple (VHS) y el virus de la varicela-zoster (VVZ) al aciclovir y la respuesta clínica a la terapia no se ha establecido en el hombre, y las pruebas de sensibilidad del virus no se han estandarizado. Los resultados de las pruebas de sensibilidad, expresados como la concentración de fármaco requerida para inhibir en un 50 % el crecimiento del virus en cultivo celular (ID50), varían mucho según el ensayo particular utilizado, el tipo de célula empleado y el laboratorio que realiza la prueba. La ID50 de aciclovir contra aislados de HSV-1 puede oscilar entre 0,02 μg/mL (reducción de placa en células Vero) y 5,9-13,5 μg/mL (reducción de placa en células de riñón de mono verde [GMK]). El ID50 contra HSV-2 varía de 0,01 a 9,9 μg/mL (reducción de placa en células Vero y GMK, respectivamente).
Usando un método de captación de colorante en células Vero, que da valores de ID50 aproximadamente de 5 a 10 veces más altos que los ensayos de reducción de placa, se examinaron 1417 aislamientos de HSV (553 HSV-1 y 864 HSV-2) de aproximadamente 500 pacientes durante un período de 5 años. período de año. Estos ensayos encontraron que el 90 % de los aislados de HSV-1 eran sensibles a ≤ 0,9 μg/mL de aciclovir y el 50 % de todos los aislados eran sensibles a ≤ 0,2 μg/mL de aciclovir. Para los aislados de HSV-2, el 90 % fue sensible a ≤ 2,2 μg/mL y el 50 % de todos los aislados fueron sensibles a ≤ 0,7 μg/mL de aciclovir. Se encontraron aislamientos con sensibilidad significativamente disminuida en 44 pacientes. Cabe destacar que ni los pacientes ni los aislados fueron seleccionados al azar y, por tanto, no representan a la población general. La mayoría de los aislamientos clínicos de HSV menos sensibles han sido relativamente deficientes en la timidina quinasa viral (TK). También se han informado cepas con alteraciones en TK viral o ADN polimerasa viral.
El ID50 contra VZV varía de 0,17 a 1,53 μg/mL (reducción del rendimiento, fibroblastos de prepucio humano) a 1,85 a 3,98 μg/mL (reducción de focos, fibroblastos de embrión humano [HEF]). La reproducción del genoma de EBV se suprime en un 50 % en células Raji superinfectadas o células linfoblastoides P3HR-1 con 1,5 μg/mL de aciclovir. El citomegalovirus (CMV) es relativamente resistente al aciclovir con valores ID50 que van desde 2,3 a 17,6 μg/mL (reducción de placa, células HEF) a 1,82 a 56,8 μg/mL (hibridación de ADN, células HEF). No se sabe que el estado latente del genoma de ninguno de los herpesvirus humanos sea sensible al aciclovir.
Resistencia
La exposición prolongada de HSV a concentraciones subinhibitorias (0.1 μg/mL) de aciclovir en cultivo celular ha resultado en la aparición de una variedad de cepas resistentes a aciclovir. Se cree que la aparición de cepas resistentes se produce por "selección" de virus naturales con susceptibilidad relativamente baja al aciclovir. Dichas cepas se han informado en aislamientos previos a la terapia de varios estudios clínicos.
Se han descrito dos mecanismos de resistencia relacionados con la timidina quinasa viral (requerida para la activación del aciclovir). Estos son: (a) selección de mutantes deficientes en timidina-quinasa que inducen poca o ninguna actividad enzimática después de la infección, y (b) selección de mutantes que poseen una timidina-quinasa de especificidad de sustrato alterada que es capaz de fosforilar el nucleósido natural timidina pero no aciclovir. La mayoría de los virus menos susceptibles que surgen in vitro son del tipo deficiente en timidina-quinasa que tienen una infectividad y patogenicidad reducidas y menos probabilidad de inducir latencia en los animales.
Sin embargo, se encontró que una infección por HSV resistente a aciclovir en un receptor de trasplante de médula ósea inmunosuprimido en terapia prolongada con aciclovir se debió a un aislado clínico que tenía una timidina quinasa normal pero una ADN polimerasa alterada. Este tercer mecanismo de resistencia que involucra a la ADN polimerasa del virus del herpes simple se debe a la selección de mutantes que codifican una enzima alterada, que es resistente a la inactivación por el trifosfato de aciclovir.
VZV parece manifestar resistencia al aciclovir a través de mecanismos similares a los observados en HSV.
Sin embargo, la investigación clínica limitada no ha revelado evidencia de un cambio significativo en la susceptibilidad in vitro del VZV con la terapia con aciclovir, aunque los mutantes resistentes de este virus pueden aislarse in vitro de manera análoga al HSV. El análisis de un pequeño número de aislados clínicos de pacientes que recibieron aciclovir oral o placebo para el herpes zoster agudo sugiere que la aparición in vivo de VZV resistente puede ocurrir con poca frecuencia. El tratamiento prolongado con aciclovir de pacientes altamente inmunocomprometidos con síndrome de inmunodeficiencia adquirida y VZV grave puede conducir a la aparición de virus resistentes.
La resistencia cruzada a otros antivirales ocurre in vitro en mutantes resistentes a aciclovir. Los mutantes del HSV que son resistentes al aciclovir debido a la ausencia de la timidina quinasa viral son resistentes a otros agentes que son fosforilados por la timidina quinasa del virus del herpes, como la bromovinildeoxiuridina, el ganciclovir y los nucleósidos 2'-fluoropirimidina, como el 2'-fluoro -5-yodoarabinosil-citosina (FIAC).
La respuesta clínica al tratamiento con aciclovir generalmente ha sido buena para los pacientes con inmunidad normal en los que se ha recuperado el HSV con sensibilidad reducida al aciclovir, ya sea antes, durante o después de la terapia. Sin embargo, se ha identificado que ciertos grupos de pacientes, como los gravemente inmunocomprometidos (especialmente los receptores de trasplante de médula ósea) y los que se someten a regímenes supresores crónicos, se asocian con mayor frecuencia con la aparición de cepas de herpes simple resistentes, que pueden o no acompañar a una respuesta deficiente. a la droga Debe reconocerse la posibilidad de aparición de virus menos sensibles al tratar a estos pacientes, y debe alentarse la vigilancia de la susceptibilidad de los aislados clínicos de estos pacientes.
En resumen, la relación cuantitativa entre la susceptibilidad in vitro de HSV y VZV al aciclovir y la respuesta clínica a la terapia no se ha establecido claramente en el hombre. Se requieren métodos estandarizados de prueba de sensibilidad del virus para permitir correlaciones más precisas entre la sensibilidad del virus in vitro y la respuesta clínica a la terapia con aciclovir.
Toxicología
Estudios de toxicidad aguda
Ratones y ratas adultos : La toxicidad aguda del aciclovir oral se determinó como se muestra en la Tabla 6.
Ratas neonatales, inmaduras y adultas
Grupos de 10 ratas Charles River CD (Sprague-Dawley) machos y 10 hembras recibieron dosis grandes únicas (5 niveles de dosis diferentes) de una solución (pH 11,0) de aciclovir mediante inyección subcutánea cuando tenían 3, 10, 28 y 71 días. de edad. Se observaron durante 14 días después del tratamiento y los valores de DL50 se calcularon mediante el método de Litchfield y Wilcoxon (ver la Tabla 7 a continuación). Este estudio se realizó para determinar si la edad de exposición afecta la toxicidad aguda del aciclovir; no hubo evidencia de que las ratas jóvenes fueran más sensibles que las ratas mayores a los efectos tóxicos agudos del aciclovir.
No hubo una relación aparente entre la duración de la supervivencia después del tratamiento y la edad a la que se administró el tratamiento. Los signos clínicos de las ratas tratadas a los 3 y 10 días de edad incluyeron ampollas cutáneas rojas y moradas, áreas azules, costras, cicatrices, piel necrótica y desprendida, heridas abiertas, temblores corporales y alopecia. En ratas tratadas con 28 y 71 días de edad.
Estudio de Toxicidad Oral Subcrónica
Ratones
Cuatro grupos, cada uno compuesto por 28 ratones Charles River CD-1 (ICR) machos y 28 hembras, recibieron dosis orales por sonda estomacal durante 33 días con suspensiones de aciclovir. Los niveles de dosis diarios fueron 0, 50, 150 y 450 mg/kg. Se realizaron mediciones hematológicas y químicas clínicas en 8 ratones macho y 8 hembras adicionales por grupo (dosificados de la misma manera) después de la primera y cuarta semana de dosificación y durante la tercera semana posterior a la dosis.
Las concentraciones de fármaco en plasma se midieron en muestras agrupadas de 4 ratones macho y 4 hembras adicionales por grupo en los días de dosis 1, 15 y 30.
Sobre la base de experimentos preliminares con ratas y ratones, se seleccionó la dosis alta de 450 mg/kg para producir los niveles plasmáticos más altos del fármaco alcanzables, de manera práctica, mediante dosificación oral en especies de roedores. Las concentraciones plasmáticas promedio del fármaco oscilaron entre aproximadamente 3,4 (a la dosis baja) y 11,0 (a la dosis alta) μg/mL de plasma una hora después de la administración oral.
No se produjeron cambios en las mediciones de salud, tasa de crecimiento, hematología y química clínica que pudieran atribuirse definitivamente a la dosificación con aciclovir. Los exámenes macroscópicos e histopatológicos de 16 ratas macho y 16 hembras de los grupos de dosis alta y de control al final del período de dosis no revelaron nada destacable.
Estudios de toxicidad crónica
Estudio de toxicidad oral de por vida en ratas que recibieron aciclovir por intubación gástrica
ratas Charles River CD (Sprague-Dawley) se les administraron suspensiones de aciclovir por sonda. Había 50 ratas macho y 50 hembras en cada uno de los siguientes niveles de dosis: 0, 50, 150 y 450 mg/kg. Después de 30 y 52 semanas de tratamiento, se necropsiaron 10 ratas macho y 10 hembras de cada grupo. Las ratas restantes recibieron dosis cada día hasta que la mortalidad natural disminuyó el tamaño del grupo a aproximadamente el 20% del número de animales de ese sexo presentes en los grupos de prueba cuando se inició el estudio. Todas las ratas restantes se sacrificaron y se les realizó la necropsia cuando se alcanzó el punto de corte del 20%. Esto fue durante la semana 110 para las ratas macho y la semana 122 para las ratas hembra. Los tejidos de las ratas de control y las del grupo de dosis alta se evaluaron mediante microscopía óptica. Los tejidos de ratas en los grupos de dosis baja y media que tenían masas, nódulos o lesiones inusuales también se examinaron mediante microscopía óptica. Los tejidos fijos de ratas que se encontraron muertas durante las primeras 52 semanas del estudio también se evaluaron mediante microscopía óptica.
No se observaron signos de toxicosis. Se recogieron muestras de plasma 1,5 horas después de la dosificación en los días 7, 90, 209, 369, 771 (solo hombres) y 852 (solo mujeres). Los niveles plasmáticos medios encontrados en machos que recibieron dosis altas (450 mg/kg/día) en los tiempos indicados anteriormente fueron los siguientes: 1,54, 1,63, 1,39, 1,60 y 1,70 μg/mL (6,84, 7,26, 6,17, 7,10 y 7,56 μM) . Los niveles plasmáticos medios correspondientes para las hembras de dosis alta para los períodos de tiempo correspondientes fueron 1,76, 2,38, 2,12, 1,71 y 1,81 μg/mL (7,82, 10,58, 9,44, 7,62 y 8,03 μM). Los niveles plasmáticos tanto en machos como en hembras en todos los niveles de dosis después de un año de tratamiento fueron generalmente comparables a los niveles plasmáticos obtenidos en muestreos anteriores. Los valores de las pruebas de laboratorio, incluidos hematología, química clínica y oftalmoscopia, estaban todos dentro del rango normal. No hubo lesiones macroscópicas o microscópicas inducidas por fármacos y no hubo evidencia de que el aciclovir afectara la supervivencia.
Estudio de carcinogenicidad oral de por vida en ratas
No hubo signos de toxicosis en ratas Charles River CD (Sprague-Dawley) (100 ratas/sexo/grupo de dosis) que recibieron aciclovir por sonda oral a 50, 150 y 450 mg/kg en un estudio de carcinogenicidad oral de por vida. Los niveles plasmáticos medios obtenidos en machos que recibieron dosis altas 1,5 horas después de la administración en varios momentos de muestreo durante el estudio fueron los siguientes: 1,54, 1,63, 1,39, 1,60 y 1,70 μg/mL (6,84, 7,26, 6,17, 7,10 y 7,56 μM) en días 7, 90, 209, 369 y 771, respectivamente. Los valores medios correspondientes para las mujeres que recibieron la dosis alta fueron 1,76, 2,38, 2,12, 1,71 y 1,81 μg/mL (7,82, 10,58, 9,44, 7,62 y 8,03 μM) en los días 7, 90, 209, 369 y 852, respectivamente.
Los valores de las pruebas de laboratorio clínico que incluyen hematología, química clínica, análisis de orina, peso corporal, consumo de alimentos y oftalmoscopía estaban todos dentro de los rangos normales. No hubo lesiones macroscópicas o microscópicas inducidas por fármacos y no hubo evidencia de que el aciclovir afectara la supervivencia, los patrones temporales de incidencia de tumores o los recuentos de tumores para neoplasias benignas o malignas.
La mayoría de las relativamente pocas ratas encontradas muertas o moribundas durante las primeras 52 semanas de este estudio sufrieron accidentes de dosificación como lo demuestran los hallazgos post mortem de perforación esofágica que causaron derrame pleural, neumonía o mediastinitis.
Estudio de carcinogenicidad oral de por vida en ratones
No hubo signos de toxicosis en ratones Charles River CD-1 (ICR) (115 ratones/sexo/grupo de dosis) que recibieron aciclovir por sonda oral a 50, 150 y 450 mg/kg/día en un estudio de carcinogenicidad oral de por vida. Los niveles plasmáticos medios obtenidos en machos que recibieron dosis altas 1,5 horas después de la administración en varios momentos de muestreo durante el estudio fueron los siguientes: 2,83, 3,17 y 1,82 μg/mL (12,59, 14,10 y 8,10 μM) en los días 90, 365 y 541, respectivamente. Los valores medios correspondientes para las hembras de dosis alta fueron 9,81, 5,85 y 4,0 μg/mL (43,60, 26,0 y 17,79 μM).
Los valores de las pruebas de laboratorio clínico que incluyen hematología, peso corporal y consumo de alimentos estaban todos dentro de los rangos normales. No hubo lesiones macroscópicas o microscópicas inducidas por fármacos. Los ratones hembra que recibieron 150 y 450 mg/kg de aciclovir sobrevivieron significativamente más que los ratones hembra de control; la supervivencia de los machos tratados fue comparable a la supervivencia de los machos de control. Los patrones de incidencia de tumores y los recuentos de tumores para neoplasias benignas o malignas no se vieron afectados por el tratamiento con aciclovir.
Estudio de toxicidad oral crónica de 12 meses en perros
Perros Beagle de raza pura recibieron 0, 15, 45 o 150 mg/kg/día de aciclovir cada día durante las dos primeras semanas de un estudio de un año. Había 9 perros machos y 9 hembras en cada grupo de prueba. Los perros recibieron cápsulas de gelatina que contenían la dosis adecuada. Se trataron tres veces al día, por lo que las dosis administradas en cada uno de los tres períodos de dosis igualmente espaciados fueron 0, 5, 15 y 50 mg/kg. Los niveles de dosis de 45 y 150 mg/kg indujeron diarrea, emesis, disminución del consumo de alimentos y pérdida de peso en perros machos y hembras durante las dos primeras semanas del estudio. Por esta razón, durante la tercera semana del estudio se tomó la decisión de disminuir los niveles de dosis media y alta a 30 y 60 mg/kg/día (10 y 20 mg/kg tid). La dosis baja de 15 mg/kg/día (5 mg/kg tid) no se modificó. Los perros que recibieron 60 mg/kg/día ocasionalmente vomitaron y ocasionalmente tuvieron diarrea, pero les fue bien durante la prueba, y los valores de aumento de peso corporal y consumo de alimentos fueron comparables a los valores de control.
Durante la toxicosis inducida por las dosis más altas de aciclovir, los niveles plasmáticos del fármaco probablemente fueron muy altos (como lo indican los valores medios iniciales de 24,0 μg/mL (106,6 μM) para los hombres que recibieron dosis altas y 17,4 μg/mL (77,2 μM) para hembras de dosis alta cuando se determina 1 hora después de la tercera dosis en el día 1 del estudio). Cuando se midió el día 15, los niveles plasmáticos de aciclovir en perros con dosis altas (150 mg/kg/día) seguían siendo muy altos, pero disminuyeron más tarde cuando se redujeron las dosis. Los valores de los niveles plasmáticos después de 12 meses de tratamiento fueron generalmente comparables a los valores registrados después de 1, 3 y 6 meses de tratamiento. Por lo tanto, no hubo indicios de un aumento del metabolismo del aciclovir como resultado del tratamiento crónico.
Durante la semana 13, algunos perros machos y hembras con niveles de dosis media y alta tuvieron los siguientes signos: sensibilidad en las patas delanteras, erosión de las almohadillas de las patas y rotura y aflojamiento de las uñas. La regeneración de las uñas perdidas comenzó unas semanas más tarde. Las uñas se regeneraron a los 6 meses (cuando se sacrificaron 3 machos y 3 hembras de cada grupo para un sacrificio intermedio) y al final del estudio eran de buena calidad en general. Nunca hubo signos de un efecto en las patas o las uñas de los perros en el grupo de dosis baja (15 mg/kg/día).
Se acepta que la lesión del epitelio corial que produce queratina ungueal puede dar como resultado una producción de queratina detenida y una producción de queratina anormal. La toxicosis transitoria inducida por las grandes dosis (45 y 150 mg/kg/día) de aciclovir administradas durante las dos primeras semanas del estudio puede haber afectado el epitelio corial. Si hubo un efecto transitorio en el epitelio corial (posiblemente relacionado con efectos directos o secundario a una enfermedad inducida por fármacos durante las dos primeras semanas del estudio), la pérdida posterior de la uña podría ser una secuela. No se observaron efectos perceptibles sobre otros tejidos productores de queratina o que contienen queratina. Cabe destacar que las alteraciones en las uñas parecieron estar relacionadas con la toxicosis transitoria inducida por los niveles de dosis de 50 y 150 mg/kg/día probados durante las dos primeras semanas del estudio y no con los de 30 y 60 mg/kg. niveles de dosis /día probados posteriormente.
No hubo alteraciones importantes inducidas por fármacos en los valores de las pruebas bioquímicas séricas, análisis de orina y pruebas electrocardiográficas realizadas a intervalos apropiados durante este estudio. Los valores de albúmina sérica y proteína total se redujeron ligeramente en perros tratados con 30 y 60 mg/kg/día durante 6 y 12 meses. Sin embargo, todos los valores de estos parámetros permanecieron dentro de los límites aceptados como normales.
Con la excepción de alteraciones residuales en la queratina vieja en las puntas de las garras, no hubo signos de efectos relacionados con el tratamiento en ninguno de los tejidos examinados por microscopía óptica. Tampoco hubo alteraciones significativas en los valores de los órganos pesados en la necropsia. Por lo tanto, los niveles de dosis de hasta 60 mg/kg/día fueron bien tolerados durante un año. El nivel de dosis “sin efecto de dosis” de aciclovir fue de 15 mg/kg/día (5 mg/kg tid); sin embargo, los únicos efectos adversos con 30 o 60 mg/kg/día fueron cambios en las uñas y las almohadillas de las patas (30 y 60 mg/kg/día) y signos gastrointestinales leves (60 mg/kg/día).
Estudios de Reproducción
Teratología – Ratas
Se administró aciclovir a ratas hembra preñadas ARS Sprague-Dawley mediante inyección subcutánea durante el período de organogénesis (del día 6 al día 15 de gestación) a niveles de dosis de 0,0, 6,0, 12,5 y 25,0 mg/kg de peso corporal dos veces al día.
Los criterios evaluados para el efecto compuesto incluyeron el peso corporal materno, el aumento de peso, la apariencia y el comportamiento, las tasas de supervivencia, los cambios en los ojos, las tasas de embarazo y los datos de reproducción. También se evaluaron la viabilidad y el desarrollo de las crías.
Además de las medidas anteriores, se sacrificaron animales designados 1 hora después de la primera dosis el día 15 para recoger muestras de sangre materna, líquido amniótico y fetos para medir la concentración del fármaco. Los valores medios de estas muestras se enumeran en la Tabla 8.
Los valores obtenidos para el plasma representarían alrededor del 30% de los niveles plasmáticos iniciales a juzgar por la vida media plasmática en roedores.
No se observaron efectos atribuibles a la administración de aciclovir en las comparaciones de valores de peso corporal materno, apariencia y comportamiento, tasas de supervivencia, tasas de embarazo o eficiencias de implantación. Además, no se observaron diferencias relacionadas con el compuesto en las evaluaciones del tamaño, sexo y desarrollo fetal.
Aunque las incidencias de reabsorción y viabilidad fetal estuvieron dentro del rango de variabilidad normal en todos los grupos, se observaron incidencias ligeramente mayores de reabsorciones en los animales que recibieron dosis altas sacrificados en los días 15 y 19 de gestación; sin embargo, no se produjeron tendencias claras relacionadas con la dosis.
Por lo tanto, el aciclovir no se consideró teratogénico o embriotóxico cuando se administró a ratas en niveles de hasta 50,0 mg/kg de peso corporal por día durante la organogénesis.
Teratología – Conejos
Se realizó un estudio de teratología en conejos blancos de Nueva Zelanda usando esencialmente el mismo diseño experimental que en la rata, excepto que la dosificación fue desde el día 6 hasta el día 18 de gestación. Además, la recolección de fetos, líquido amniótico y muestras de sangre materna se realizó el día 18 en lugar del día 15.
No se observaron signos de toxicidad materna con ninguna dosis, pero hubo una menor eficiencia de implantación estadísticamente significativa (p
Se detectaron concentraciones de aciclovir en muestras de plasma y líquido amniótico, así como en homogeneizados de tejidos fetales. Todas las muestras se tomaron una hora después de la primera dosis el día 18 de gestación. Las concentraciones de fármaco en el líquido amniótico fueron sustancialmente más altas que las del plasma (ver Tabla 9).
Reproducción – Fertilidad
Se demostró que el aciclovir no afecta la fertilidad ni la reproducción en grupos de 15 ratones machos y 30 hembras en un estudio de fertilidad de dos generaciones. Los ratones de este estudio recibieron aciclovir mediante intubación gástrica en dosis de 50, 150 y 450 mg/kg/día. Los machos recibieron la dosis durante 64 días consecutivos antes del apareamiento y las hembras durante 21 días antes del apareamiento.
En un estudio de fertilidad en ratas en el que grupos de 20 ratas macho y 20 hembras recibieron 0, 12,5, 25,0 y 50,0 mg/kg/día mediante inyección subcutánea, se demostró que el aciclovir no tiene ningún efecto sobre el apareamiento o la fertilidad. Los machos recibieron la dosis durante 60 días antes del apareamiento y hasta que se completó su horario de apareamiento. Se administró la dosis a ratas hembra durante 14 días antes del apareamiento y hasta el día 7 de gestación. Con 50 mg/kg/día sc hubo un aumento estadísticamente significativo en la pérdida posterior a la implantación, pero no una disminución concomitante en el tamaño de la camada.
En 25 conejas tratadas por vía subcutánea con 50 mg/kg/día de aciclovir en los días 6 a 18 de gestación, hubo una disminución estadísticamente significativa en la eficiencia de implantación pero no una disminución concomitante en el tamaño de la camada. También hubo un aumento relacionado con la dosis en el número de fetos con costillas supernumerarias en todos los grupos tratados con el fármaco. Este aumento no estuvo relacionado con la dosis cuando se examinó la incidencia de costillas supernumerarias por camada.
En 15 conejas tratadas por vía intravenosa con 50 mg/kg/día de aciclovir en los días 6 a 18 de gestación, no hubo efecto sobre la eficiencia de implantación ni sobre el tamaño de la camada.
En un estudio perinatal y posnatal en ratas (20 ratas hembra por grupo), se administró aciclovir por vía subcutánea en dosis de 0, 12,5, 25 y 50 mg/kg/día desde los 17 días de gestación hasta los 21 días posparto. Con 50 mg/kg/día sc hubo una disminución estadísticamente significativa en el número medio del grupo de cuerpos lúteos, sitios de implantación totales y fetos vivos en la generación F1. Aunque no fue estadísticamente significativo, también hubo una disminución relacionada con la dosis en el número medio de fetos vivos y sitios de implantación del grupo con 12,5 mg/kg/día y 25 mg/kg/día sc
En un estudio de búsqueda de rango de dosis con 5 conejas, la administración intravenosa de aciclovir a una dosis de 100 mg/kg/día desde el día 6 al 8 de embarazo, una dosis conocida por causar nefropatía obstructiva, provocó un aumento significativo en las reabsorciones fetales y una disminución correspondiente en el tamaño de la camada. A una dosis intravenosa máxima tolerada de 50 mg/kg/día en conejos, no hubo efectos reproductivos relacionados con el fármaco.
En un estudio de toxicidad subcrónica en el que se administraron dosis intraperitoneales de aciclovir de 0, 20, 80 ó 320 mg/kg/día durante un mes a grupos de 20 ratas macho y 20 hembras, y se les dio seguimiento durante un período de un mes posterior a la dosis, hubo atrofia. Alguna evidencia histológica de recuperación de la producción de esperma fue evidente 30 días después de la dosis, pero este fue un tiempo insuficiente para demostrar la reversibilidad total.
grupos de 25 ratas macho y 25 hembras se les administraron dosis intraperitoneales de aciclovir a 0, 5, 20 u 80 mg/kg/día durante 6 meses. Diez ratas macho y 10 hembras de cada grupo se continuaron sin dosificar durante 13 semanas. La atrofia testicular se limitó a ratas que recibieron dosis altas de 80 mg/kg/día durante 6 meses. Los datos de peso de los órganos y la microscopía óptica definieron la reversibilidad total de la atrofia testicular al final del período de recuperación posterior a la dosis.
En un estudio con perros de 31 días (16 machos y 16 hembras por grupo) en el que se administró aciclovir por vía intravenosa a niveles de 50, 100 y 200 mg/kg/día, los testículos de los perros eran normales a 50 mg/kg. Dosis de 100 ó 200 mg/kg/día causaron la muerte de algunos perros por efectos citostáticos (médula ósea y epitelio gastrointestinal) y testículos aspérmicos o testículos con túbulos aspérmicos dispersos. No se puede descartar que el cambio testicular haya sido primario, sin embargo, se pueden observar cambios similares secundarios al estrés severo en perros moribundos.
Estudios de toxicidad del desarrollo
Ratas Neonatales - Estudio Subcrónico
Se administró aciclovir disuelto en solución salina estéril al 0,4 % mediante inyección subcutánea a ratas recién nacidas Charles River CD (Sprague-Dawley) durante 19 días consecutivos, comenzando el tercer día posparto. Los niveles de dosis probados fueron 0, 5, 20 y 80 mg/kg de peso corporal. Hubo 12 camadas (cada una compuesta por 5 neonatos machos y 5 hembras amamantando a la madre natural) en cada nivel de dosis. Las presas no fueron tratadas. Se extrajo a los recién nacidos de cada grupo para realizar la necropsia y la evaluación microscópica de una amplia variedad de tejidos, incluidos los ojos y múltiples secciones del cerebro, después de haber sido tratados durante 5, 12 o 19 días y después de un período de 3 semanas sin el fármaco después de la dosis ( en cuyo momento tenían 45 días de edad). Se realizaron pruebas hematológicas (hemoglobina, volumen de células empaquetadas, glóbulos rojos, glóbulos blancos y recuento diferencial de células) y química clínica (BUN) después de 16 días de tratamiento y se repitieron 18 días después de que se administró la última dosis (19).
Se extrajo sangre de algunos neonatos 30 minutos después del tratamiento el día 1, el día 9 y al final del período de dosificación para la determinación de las concentraciones de aciclovir en plasma. La mayor concentración de aciclovir en plasma fue de 99,1 μg/mL (440,5 μM) encontrada en el plasma agrupado recolectado de 6 neonatos femeninos que recibieron una dosis alta (80 mg/kg) 30 minutos después de administrar la primera dosis. El tratamiento con aciclovir no aumentó la mortalidad en el período neonatal.
Las ratas en el grupo de dosis baja ganaron tanto peso corporal como las ratas de control respectivas. Se observaron reducciones significativas (p
Los exámenes oculares y el microscopio óptico no revelaron efectos adversos sobre el desarrollo ocular. Debe enfatizarse que no hubo evidencia morfológica o funcional de efectos adversos en el cerebro en desarrollo u otras partes del sistema nervioso central. Por lo tanto, el aciclovir es claramente diferente del arabinósido de citosina que, según se informó, produce displasia cerebelosa y retiniana prominente en ratas recién nacidas.
Mutagenicidad y otros estudios a corto plazo
Se ha probado el potencial mutagénico del aciclovir en varios sistemas in vitro e in vivo: 10 de noviembre de 2014 Página 27 de 38
Microbiano
Se probó la actividad mutagénica de aciclovir en el ensayo de placa Ames Salmonella; en una modificación de preincubación del ensayo de Ames; en el ensayo de reparación de ADN Rosenkrantz E. coli polA+/polA-; y en la eucariota S. cerevisiae, D-4. Todos los estudios se realizaron tanto en presencia como en ausencia de activación metabólica exógena de mamíferos. Aciclovir no dio respuestas positivas en ninguno de estos sistemas.
Los estudios previos de Salmonella se extendieron a concentraciones extremadamente altas para lograr toxicidad. No se observaron efectos positivos ni en presencia ni en ausencia de activación metabólica exógena en mamíferos, a concentraciones de aciclovir de hasta 300 mg/placa u 80 mg/ml.
Sistemas de mamíferos
Se analizó la actividad mutagénica del aciclovir en células de linfoma de ratón L5178Y cultivadas, heterocigotas en el locus de la timidina quinasa (TK), midiendo la tasa de mutación directa a la deficiencia de TK (TK+/- → TK-/-; se realizaron estudios adicionales en el HGPRT locus y en el marcador de resistencia a la uabaína en estas mismas células. Todos los estudios se realizaron en presencia y en ausencia de activación metabólica exógena de mamíferos. El compuesto de prueba fue mutagénico en el locus TK a altas concentraciones (400 -2400 μg/mL) (En comparación, el límite superior de los niveles plasmáticos máximos de aciclovir después de una dosis oral de 200 mg cada 4 horas es de 0,9 μg/mL). Fue negativo en el locus HGPRT y en el marcador de resistencia a la uabaína. Se obtuvieron resultados idénticos con y sin activación metabólica.
Se obtuvieron resultados no concluyentes sin una respuesta aparente relacionada con la dosis cuando se estudió la mutagenicidad del aciclovir en cada uno de los 3 loci (APRT, HGPRT y resistencia a la uabaína) en células de ovario de hámster chino (CHO), tanto en presencia como en ausencia de activación metabólica exógena.
Se ha demostrado que el aciclovir, a una concentración de 50 μg/mL (222 μM) para una exposición de 72 horas, causa un aumento estadísticamente significativo en la incidencia de focos transformados morfológicamente como resultado del tratamiento de células BALB/C-3T3 in vitro en la ausencia de activación metabólica exógena. Se ha demostrado que los focos transformados morfológicamente crecen como tumores después del trasplante en ratones destetados, singénicos e inmunodeprimidos. Los tejidos tumorales se diagnosticaron como sarcomas indiferenciados o linfosarcomas.
El aciclovir, en concentraciones entre 8 y 64 μg/mL durante 18 horas de exposición, no indujo ningún foco transformado morfológicamente entre las células C3H/10T ½ tratadas in vitro en ausencia de activación metabólica exógena.
El aciclovir, en concentraciones de 62,5 y 125 μg/mL para una exposición de 48 horas, no indujo ninguna aberración cromosómica en linfocitos humanos cultivados en ausencia de activación metabólica exógena. A concentraciones más altas, 250 y 500 μg/mL durante 48 horas de exposición, el aciclovir provocó un aumento significativo en la incidencia de rotura cromosómica. También hubo una disminución significativa relacionada con la dosis en el índice mitótico con la exposición al aciclovir.
El aciclovir, a dosis de 25 y 50 mg/kg/día ip durante 5 días consecutivos, no produjo un efecto letal dominante en ratones macho BKA (CPLP). Además, no hubo evidencia de un efecto letal dominante en ratones machos y hembras Charles River CD-1 (ICR) tratados por vía oral a niveles de dosis de 50, 150 y 450 mg/kg/día como se resume en el Estudio de reproducción/fertilidad de dos generaciones. .
El aciclovir, en dosis intraperitoneales únicas de 25, 50 y 100 mg/kg, no logró inducir aberraciones cromosómicas en las células de la médula ósea de los hámsters chinos cuando se examinaron 24 horas después de la administración. A dosis nefrotóxicas más altas (500 y 1000 mg/kg), se observó un efecto blastogénico. (Una dosis intraperitoneal de 500 mg/kg produce niveles plasmáticos máximos medios en hámsteres chinos de 611 μg/mL (2,72 mM), que es 680 veces mayor que el límite superior de los niveles plasmáticos máximos en humanos durante la dosificación oral de 200 mg q4h).
El aciclovir, en dosis intravenosas únicas de 25, 50 y 100 mg/kg, no logró inducir aberraciones cromosómicas en las células de la médula ósea de ratas macho y hembra cuando se examinaron a las 6, 24 y 48 horas después del tratamiento.
Por lo tanto, todos estos estudios demostraron que el aciclovir no causa mutaciones de un solo gen sino que es capaz de romper los cromosomas.
Estudios de Inmunotoxicología
El aciclovir se sometió a una serie de pruebas inmunológicas in vitro e in vivo.
En dos pruebas in vivo, citotoxicidad mediada por linfocitos y quimiotaxis de neutrófilos, el aciclovir no mostró efectos inhibitorios en concentraciones tan altas como 135 μg/mL (600 μM). El compuesto inhibió la formación de rosetas en aproximadamente un 50 % a 0,9 μg/mL (4 μM).
En cuatro pruebas in vivo en ratones que midieron la inmunidad mediada por células (citotoxicidad celular dependiente del complemento, citotoxicidad celular independiente del complemento, hipersensibilidad retardada y reacción injerto contra huésped), el aciclovir no mostró efectos inhibitorios en dosis únicas de hasta 200 mg/kg administradas el día 2 después de la estimulación antigénica.
Cuatro dosis diarias de 100 mg/kg/día no tuvieron un efecto significativo sobre las placas de hemolisina de Jerne o los anticuerpos circulantes el día 7 después de la estimulación antigénica. Cuando se examinaron las placas de hemolisina de Jerne y los títulos de anticuerpos cuatro días después de la exposición antigénica y un día después de la última dosis de fármaco, 100 mg/kg mostraron sólo un ligero efecto supresor. Sin embargo, 200 mg/kg produjeron cierta pérdida de peso (-2,2 g), una reducción moderada en el número de placas de hemolisina de Jerne (PFC/bazo se redujo al 33% del control, PFC/107 WBC al 46,5% del control). Sin embargo, solo hubo una pequeña reducción en el título de hemaglutinina circulante (de 8,3 a 6,5) y el título de hemolisina circulante (de 9,5 a 8,3) con 200 mg/kg.
En experimentos en ratones diseñados para probar si el aciclovir potenciaría el efecto inmunosupresor de la azatioprina sobre la formación de anticuerpos, se encontró que los efectos de los dos fármacos no eran más que aditivos. Solo la dosis de 200 mg/kg de aciclovir mostró una mayor supresión de la respuesta de anticuerpos cuando se administró en combinación con azatioprina en dosis superiores a 25 mg/kg.
Se llevaron a cabo estudios para evaluar la influencia del aciclovir in vitro sobre la función de los linfocitos humanos. Los efectos inhibitorios sobre la blastogénesis se observaron solo en ensayos que examinaron concentraciones máximas de mitógenos potentes, fitohemaglutinina (PHA) y concanavalina A (Con A), y solo a concentraciones de fármaco superiores a 50 μg/mL (222 μM) y fueron mucho menores con monilia y antígenos del toxoide tetánico, donde la respuesta blastogénica es característicamente menos vigorosa. Hubo muy poco efecto sobre la citotoxicidad o la producción de LIF excepto a concentraciones de 200 μg/mL (890 μM) donde ya se ha demostrado que existe un efecto citotóxico directo. Estas concentraciones inhibidoras superan con creces los niveles anticipados de las dosis seleccionadas para la aplicación clínica y son más de 1000 veces superiores a la concentración requerida para inhibir la multiplicación del herpesvirus in vitro.
Se midió el efecto del aciclovir sobre células humanas. Una concentración de 11,2 - 22,5 μg/mL (50-100 μM) inhibe la división de los fibroblastos en grado variable, según el diseño experimental y la confluencia de la monocapa. La magnitud de este efecto fue menor que la causada por el arabinósido de adenina o el interferón leucocitario humano cuando se compararon estos tres agentes antivirales en concentraciones clínicamente relevantes. El aciclovir también inhibió la incorporación de timidina por parte de las células mononucleares de sangre periférica estimuladas por PHA o tres antígenos de herpesvirus diferentes. Se observó una curva dosis-respuesta lineal con estas células y su proliferación se inhibió en un 50 % con 22,5 μg/mL (100 μM) de aciclovir. Se ejerció inhibición sobre la proliferación de células T sin efecto aparente sobre la liberación de linfocinas o sobre la función de los monocitos.
También se debe mencionar que no hubo evidencia de efectos adversos sobre el sistema inmunológico en las pruebas detalladas subcrónicas y crónicas en animales cubiertas anteriormente en este resumen, excepto a dosis excesivamente altas (50 a 100 mg/kg dos veces al día) en perros en los que se observó hipoplasia linfoide marcada. ocurrió.
REFERENCIAS
1. Balfour HH, Jr., Kelly JM, Suarez CS, Heussner RC, Englund JA, Crane DD et al. Tratamiento con aciclovir de la varicela en niños por lo demás sanos. J Pediatr 1990; 116(4):633-639.
2. Balfour HH, Jr., Rotbart HA, Feldman S, Dunkle LM, Feder HM, Jr., Prober CG et al. Tratamiento con aciclovir de la varicela en adolescentes por lo demás sanos. El Grupo de Estudio Colaborativo de Aciclovir Varicela. J Pediatr 1992; 120 (4 punto 1): 627-633.
3. Barry DW, Blum SR. Fármacos antivirales: aciclovir, en Recent Advances in Clinical Pharmacology. Turner P, Shand DG (eds) Churchill Livingstone, Edimburgo 1983.
4. Barry DW, Nusinoff-Lehrman S. Resistencia viral en la práctica clínica: resumen de cinco años de experiencia con aciclovir. Enfoques farmacológicos y clínicos de los herpesvirus y la quimioterapia viral, Aiso, Japón, 10 al 13 de septiembre de 1984.
5. Barry DW, Nusinoff-Lehrman S, Ellis MN, Biron KK, Furman PA. Resistencia viral, experiencia clínica. Scand J Infect Dis Suppl 1985; 47:155-164.
6. Barry DW, Nusinoff-Lehrman S. Resistencia viral en la práctica clínica: resumen de cinco años de experiencia con aciclovir. Actas del Simposio internacional sobre enfoques farmacológicos y clínicos de los virus del herpes y la quimioterapia viral, Elsever, Amsterdam 1985;269-270.
7. Biron KK, Elion GB. Efecto del aciclovir combinado con otros agentes antiherpéticos sobre el virus varicela zoster in vitro. Am J Med 1982; 73(1A):54-57.
8. Boelaert J, Schurgers M, Daneels R, Van Landuyt HW, Weatherley BC. Farmacocinética de dosis múltiples de aciclovir intravenoso en pacientes en diálisis peritoneal ambulatoria continua. J Antimicrob Chemother 1987; 20(1):69-76.
9. Bryson YJ, Dillon M, Lovett M, Acuna G, Taylor S, Cherry JD et al. Tratamiento de los primeros episodios de infección por el virus del herpes simple genital con aciclovir oral. Un ensayo controlado aleatorio doble ciego en sujetos normales. N Engl J Med 1983; 308(16):916-921.
10. Burns WH, Saral R, Santos GW, Laskin OL, Lietman PS, McLaren C et al. Aislamiento y caracterización del virus Herpes simplex resistente después de la terapia con aciclovir. Lancet 1982; 1(8269):421-423.
11. Christophers J, Sutton RN. Caracterización de aislamientos clínicos del virus del herpes simple resistentes y sensibles al aciclovir de un paciente inmunocomprometido. J Antimicrob Chemother 1987; 20(3):389-398.
12. Cole NL, Balfour HH, Jr. El virus Varicella-Zoster no se vuelve más resistente al aciclovir durante la terapia. J Infect Dis 1986; 153(3):605-608.
13. Collins P, Bauer DJ. La actividad in vitro contra el virus del herpes de 9-(2-hidroxietoximetil)guanina (acicloguanosina), un nuevo agente antiviral. J Antimicrob Chemother 1979; 5(4):431-436.
14. Collins P, Oliver NM. Vigilancia de la sensibilidad de aislados del virus del herpes simple de pacientes que reciben aciclovir. J Antimicrob Chemother 1986; 18 Suplemento B: 103-112.
15. Collins P. Sensibilidad viral después de la introducción de aciclovir. Am J Med 1988; 85(2A):129-134.
16. Collins P, Larder BA, Oliver NM, Kemp S, Smith IW, Darby G. Caracterización de un mutante de ADN polimerasa del virus del herpes simple de un paciente gravemente inmunocomprometido que recibe aciclovir. J Gen Virol 1989; 70 (Pt 2):375-382.
17. Crumpacker CS, Schnipper LE, Zaia JA, Levin MJ. Inhibición del crecimiento por acicloguanosina de herpesvirus aislados de infecciones humanas. Agentes antimicrobianos Chemother 1979; 15(5):642-645.
18. Crumpacker CS, Schnipper LE, Marlowe SI, Kowalsky PN, Hershey BJ, Levin MJ. Resistencia a fármacos antivirales del virus del herpes simple aislado de un paciente tratado con aciclovir. N Engl J Med 1982; 306(6):343-346.
19. Darby G, Inglis MM, Larder BA. Mecanismos de resistencia a los inhibidores análogos de nucleósidos del virus del herpes simple. 6th Int Congr Virol 1984; (Resumen #W34-5).
20. De Clercq E, Descamps J, Verhelst G, Walker RT, Jones AS, Torrence PF et al. Eficacia comparativa de los fármacos antiherpes frente a diferentes cepas del virus del herpes simple. J Infect Dis 1980; 141(5):563-574.
21. De Clercq E. Eficacia comparativa de fármacos antiherpes en diferentes líneas celulares. Agentes antimicrobianos Chemother 1982; 21(4):661-663.
22. Dekker C, Ellis MN, McLaren C, Hunter G, Rogers J, Barry DW. Resistencia viral en la práctica clínica. J Antimicrob Chemother 1983; 12 Suplemento B:137-152.
23. Douglas JM, Davis LG, Remington ML, Paulsen CA, Perrin EB, Goodman P et al. Un ensayo doble ciego controlado con placebo sobre el efecto del aciclovir oral administrado crónicamente sobre la producción de esperma en hombres con herpes genital frecuentemente recurrente. J Infect Dis 1988 marzo; 157:588-93.
24. Douglas JM, Critchlow C, Benedetti J, Mertz GJ, Connor JD, Hintz MA et al. Un estudio doble ciego de aciclovir oral para la supresión de las recurrencias de la infección por el virus del herpes simple genital. N Engl J Med 1984; 310(24):1551-1556.
25. Dunkle LM, Arvin AM, Whitley RJ, Rotbart HA, Feder HM, Jr., Feldman S et al. Un ensayo controlado de aciclovir para la varicela en niños normales. N Engl J Med 1991; 325(22):1539-1544.
26. Ellis MN, Keller PM, Martin JL, Strauss SE, Nusinoff-Lehrman S etal. Caracterización de un aislado clínico de HSV-2 que contiene un mutante resistente a ACV que produce una timidina quinasa con especificidad de sustrato alterada. Noveno Taller Internacional sobre Herpesvirus, Seattle, Washington, 24-29 de agosto de 1984.
27. Ellis MN, Keller PM, Fyfe JA, Martin JL, Rooney JF, Straus SE et al. Aislado clínico del virus del herpes simple tipo 2 que induce una timidina quinasa con especificidad de sustrato alterada. Agentes antimicrobianos Chemother 1987; 31(7):1117-1125.
28. Englund JA, Zimmerman ME, Swierkosz EM, Goodman JL, Scholl DR, Balfour HH, Jr. Virus del herpes simple resistente al aciclovir. Un estudio en un centro de tercer nivel de atención. Ann Intern Med 1990; 112(6):416-422.
29. Erlich KS, Jacobson MA, Koehler JE, Follansbee SE, Drennan DP, Gooze L et al. Tratamiento con foscarnet para infecciones graves por el virus del herpes simple tipo 2 resistente al aciclovir en pacientes con síndrome de inmunodeficiencia adquirida (SIDA). Un juicio sin control. Ann Intern Med 1989; 110(9):710-713.
30. Erlich KS, Mills J, Chatis P, Mertz GJ, Busch DF, Follansbee SE et al. Infecciones por el virus del herpes simple resistente al aciclovir en pacientes con síndrome de inmunodeficiencia adquirida. N Engl J Med 1989; 320(5):293-296.
31. Field HJ, Darby G, Wildy P. Aislamiento y caracterización de mutantes resistentes al aciclovir del virus del herpes simple. J Gen Virol 1980; 49(1):115-124.
32. Campo HJ. El problema de la resistencia viral inducida por fármacos, en Problemas de la terapia antiviral. Stuart-Harris CH, Oxford J (Eds) Academic Press, Londres 1983.
33. Fyfe K. Patrones de recurrencia del herpes genital después del cese de más de 5 años de supresión crónica con aciclovir. VIII Int Conf AIDS/III Std World Cong 1992; (B240).
34. Huff JC, Bean B, Balfour HH, Jr., Laskin OL, Connor JD, Corey L et al. Terapia del herpes zoster con aciclovir oral. Am J Med 1988; 85(2A):84-89.
35. Jacobson MA, Berger TG, Fikrig S, Becherer P, Moohr JW, Stanat SC et al. Infección por el virus de la varicela zoster resistente a aciclovir después de la terapia oral crónica con aciclovir en pacientes con síndrome de inmunodeficiencia adquirida (SIDA). Ann Intern Med 1990; 112(3):187-191.
36. Kaplowitz LG, Baker D, Gelb L, Blythe J, Hale R, Frost P et al. Tratamiento continuo prolongado con aciclovir de adultos normales con infección por el virus del herpes simple genital frecuentemente recurrente. El Grupo de Estudio de Aciclovir. JAMA 1991; 265(6):747-751.
37. Krasny HC, Liao SH, de Miranda P, Laskin OL, Whelton A, Lietman PS. Influencia de la hemodiálisis en la farmacocinética de aciclovir en pacientes con insuficiencia renal crónica. Am J Med 1982; 73(1A):202-204.
38. Kurtz T. Seguridad y eficacia del tratamiento supresor a largo plazo con ciclovir del herpes genital frecuentemente recurrente: resultados del año 5. 30th Intersci Conf Antimicrob Agents Chemother 1990;270.
39. Laskin OL, Longstreth JA, Whelton A, Krasny HC, Keeney RE, Rocco L et al. Efecto de la insuficiencia renal sobre la farmacocinética del aciclovir. Am J Med 1982; 73(1A):197-201.
40. Lau RJ, Emery MG, Galinsky RE. Acumulación inesperada de aciclovir en la leche materna con estimación de exposición infantil. Obstet Gynecol 1987; 69 (3 punto 2): 468-471.
41. Lehrman SN, Douglas JM, Corey L, Barry DW. Herpes genital recurrente y tratamiento supresor con aciclovir oral. Relación entre el resultado clínico y la sensibilidad al fármaco in vitro. Ann Intern Med 1986; 104(6):786-790.
42. Marlowe S, Douglas J, Corey L, Schnipper L, Crumpacker C. Sensibilidad de los aislados genitales del HSV después del aciclovir oral. 24th Interscience Conf Antimicrob Ag Chemother, Washington, DC, 8-10 de octubre de 1984.
43. Mattison HR, Reichman RC, Benedetti J, Bolgiano D, Davis LG, Bailey-Farchione A et al. Ensayo doble ciego controlado con placebo que compara el tratamiento supresor a largo plazo con el aciclovir oral a corto plazo para el tratamiento del herpes genital recurrente. Am J Med 1988; 85(2A):20-25.
44. McLaren C, Sibrack CD, Barry DW. Espectro de sensibilidad del aciclovir de aislados clínicos del virus del herpes simple. Am J Med 1982; 73(1A):376-379.
45. McLaren C, Ellis MN, Hunter GA. Un ensayo colorimétrico para la medición de la sensibilidad de los virus del herpes simple a los agentes antivirales. Antiviral Res 1983; 3(4):223-234.
46. McLaren C, Corey L, Dekket C, Barry DW. Sensibilidad in vitro al aciclovir en virus del herpes simple genital de pacientes tratados con aciclovir. J Infect Dis 1983; 148(5):868-875.
47. Mertz GJ, Critchlow CW, Benedetti J, Reichman RC, Dolin R, Connor J et al. Ensayo doble ciego controlado con placebo de aciclovir oral en el primer episodio de infección genital por el virus del herpes simple. JAMA 1984; 252(9):1147-1151.
48. Mertz GJ, Jones CC, Mills J, Fife KH, Lemon SM, Stapleton JT et al. Supresión a largo plazo con aciclovir de la infección por el virus del herpes simple genital frecuentemente recurrente. Un ensayo doble ciego multicéntrico. JAMA 1988; 260(2):201-206.
49. Mertz GJ, Eron L, Kaufman R, Goldberg L, Raab B, Conant M et al. Tratamiento prolongado con aciclovir oral continuo versus intermitente en adultos normales con infección por el virus del herpes simple genital frecuentemente recurrente. Am J Med 1988; 85(2A):14-19.
50. Meyer LJ, de Miranda P, Sheth N, Spruance S. Aciclovir en la leche materna humana. Am J Obstet Gynecol 1988; 158 (3 punto 1): 586-588.
51. Mindel A, Weller IV, Faherty A, Sutherland S, Hindley D, Fiddian AP et al. Aciclovir oral profiláctico en herpes genital recurrente. Lancet 1984; 2(8394):57-59.
52. Morton P, Thomson AN. Aciclovir oral en el tratamiento del herpes zoster en la práctica general. NZ Med J 1989; 102(863):93-95.
53. Naib ZM, Nahmias AJ, Josey WE, Zaki SA. Relación de la citohistopatología de la infección por herpesvirus genital con la anaplasia cervical. Cáncer Res 1973; 33(6):1452-1463.
54. Nilsen AE, Aasen T, Halsos AM, Kinge BR, Tjotta EA, Wikstrom K et al. Eficacia del aciclovir oral en el tratamiento del herpes genital inicial y recurrente. Lancet 1982; 2(8298):571-573.
55. Nusinoff-Lehrman S, Hunter G, Rogers J, Corey L, Davis G. La sensibilidad in vitro al aciclovir del herpesvirus arrojada por pacientes que reciben terapia oral supresiva. 24th Interscience Conf Antimicrob Ag Chemother, Washington, DC, 8-10 de octubre de 1984; (Abstract #1015).
56. O'Brien JJ, Campoli-Richards DM. Aciclovir. Una revisión actualizada de su actividad antiviral, propiedades farmacocinéticas y eficacia terapéutica. Drogas 1989; 37(3):233-309.
57. Pahwa S, Biron K, Lim W, Swenson P, Kaplan MH, Sadick N et al. Infección continua por varicela-zoster asociada con resistencia al aciclovir en un niño con sida. JAMA 1988; 260(19):2879-2882.
58. Parker AC, Craig JI, Collins P, Oliver N, Smith I. Infección por el virus del herpes simple resistente al aciclovir debido a la alteración de la ADN polimerasa. Lancet 1987; 2(8573):1461.
59. Parris DS, Harrington JE. Existen variantes del virus del herpes simple limitadas a altas concentraciones de aciclovir en aislados clínicos. Agentes antimicrobianos Chemother 1982; 22(1):71-77.
60. Preblud SR, Arbeter AM, Proctor EA, Starr SE, Plotkin SA. Susceptibilidad de las cepas vacunales del virus varicela-zoster a los compuestos antivirales. Agentes antimicrobianos Chemother 1984; 25(4):417-421.
61. Reichman RC, Badger GJ, Mertz GJ, Corey L, Richman DD, Connor JD et al. Tratamiento de infecciones genitales recurrentes por herpes simple con aciclovir oral. Un ensayo controlado. JAMA 1984; 251(16):2103-2107.
62. Shah GM, Winer RL, Krasny HC. Farmacocinética de aciclovir en un paciente en diálisis peritoneal ambulatoria continua. Am J Kidney Dis 1986; 7(6):507-510.
63. Sibrack CD, Gutman LT, Wilfert CM, McLaren C, St Clair MH, Keller PM et al. Patogenicidad del virus del herpes simple tipo 1 resistente a aciclovir de un niño inmunodeficiente. J Infect Dis 1982; 146(5):673-682.
64. Straus SE, Seidlin M, Takiff H, Jacobs D, Bowen D, Smith HA. Aciclovir oral para suprimir las infecciones recurrentes por el virus del herpes simple en pacientes inmunodeficientes. Ann Intern Med 1984; 100(4):522-524.
65. Straus SE, Takiff HE, Seidlin M, Bachrach S, Lininger L, DiGiovanna JJ et al. Supresión del herpes genital frecuentemente recurrente. Un ensayo doble ciego controlado con placebo de aciclovir oral. N Engl J Med 1984; 310(24):1545-1550.
66. Straus SE, Croen KD, Sawyer MH, Freifeld AG, Felser JM, Dale JK et al. Supresión con aciclovir del herpes genital frecuentemente recurrente. Eficacia y disminución de la necesidad durante los sucesivos años de tratamiento. JAMA 1988; 260(15):2227-2230.
67. Vinckier F, Boogaerts M, De Clerck D, De Clercq E. Infección herpética crónica en un paciente inmunocomprometido: reporte de un caso. J Oral Maxillofac Surg 1987; 45(8):723-728.
68. Wade JC, Newton B, McLaren C, Flournoy N, Keeney RE, Meyers JD. Aciclovir intravenoso para tratar la infección mucocutánea por el virus del herpes simple después del trasplante de médula: un ensayo doble ciego. Ann Intern Med 1982; 96(3):265-269.
69. Wade JC, McLaren C, Meyers JD. Frecuencia e importancia del virus del herpes simple resistente al aciclovir aislado de pacientes con trasplante de médula que reciben múltiples cursos de tratamiento con aciclovir. J Infect Dis 1983; 148(6):1077-1082.
INFORMACIÓN DEL PACIENTE
Se indica a los pacientes que consulten con su médico si experimentan reacciones adversas graves o molestas, si quedan embarazadas o tienen la intención de quedar embarazadas, si tienen la intención de amamantar mientras toman ZOVIRAX (aciclovir) administrado por vía oral o si tienen alguna otra pregunta.
Se debe advertir a los pacientes que mantengan una hidratación adecuada.