Neurontin 100mg, 300mg, 400mg, 600mg Gabapentin Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Neurontin y cómo se usa?
Neurontin 400 mg es un medicamento recetado que se usa para tratar los síntomas del dolor nervioso y las convulsiones. Neurontin puede usarse solo o con otros medicamentos.
Neurontin es un análogo de GABA, fármaco antiepiléptico.
No se sabe si Neurontin es seguro y efectivo en niños menores de 3 años.
¿Cuáles son los posibles efectos secundarios de Neurontin 400 mg?
Neurontin puede causar efectos secundarios graves que incluyen:
- aumento de las convulsiones
- debilidad severa o cansancio
- problemas con el equilibrio o el movimiento muscular
- dolor de estomago superior
- Dolor de pecho
- tos nueva o que empeora con fiebre
- dificultad para respirar
- hormigueo o entumecimiento intenso
- movimiento rápido de ojos
- orinar poco o nada
- dolor o dificultad para orinar
- hinchazón en los pies o los tobillos
Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Neurontin incluyen:
- dolor de cabeza
- mareo
- somnolencia
- cansancio
- hinchazón en las manos o los pies
- problemas con tus ojos
- problemas de coordinación
- Los efectos secundarios más comunes de Neurontin 600 mg en niños incluyen:
- fiebre
- náuseas
- vómitos
- cambios en el comportamiento
- problemas de memoria
- problemas para concentrarse
- actuando inquieto, hostil o agresivo
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Neurontin. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
DESCRIPCIÓN
El ingrediente activo de las cápsulas, comprimidos y solución oral de NEURONTIN es la gabapentina, cuyo nombre químico es ácido 1-(aminometil)ciclohexanoacético.
La fórmula molecular de la gabapentina es C9H17NO2 y el peso molecular es 171,24. La fórmula estructural de la gabapentina es:
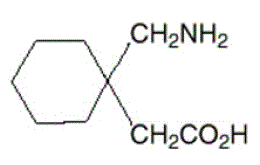
La gabapentina es un sólido cristalino de color blanco a blanquecino con un pKa1 de 3,7 y un pKa2 de 10,7. Es libremente soluble en agua y en soluciones acuosas tanto básicas como ácidas. El logaritmo del coeficiente de reparto (noctanol/tampón de fosfato 0,05 M) a pH 7,4 es –1,25.
Cada cápsula de Neurontin de 100 mg contiene 100 mg, 300 mg o 400 mg de gabapentina y los siguientes ingredientes inactivos: lactosa, almidón de maíz, talco, gelatina, dióxido de titanio, FD&C Blue No. 2, óxido de hierro amarillo (solo 300 mg y 400 mg) y óxido de hierro rojo (solo 400 mg).
Cada comprimido de Neurontin contiene 600 mg u 800 mg de gabapentina y los siguientes ingredientes inactivos: poloxámero 407, copovidona, maicena, estearato de magnesio, hidroxipropilcelulosa, talco y cera de candelilla
La solución oral de Neurontin contiene 250 mg de gabapentina por 5 ml (50 mg por ml) y los siguientes ingredientes inactivos: glicerina, xilitol, agua purificada y sabor a anís de fresa fresco artificial.
INDICACIONES
NEURONTIN® está indicado para:
- Manejo de la neuralgia posherpética en adultos
- Terapia adyuvante en el tratamiento de las crisis de inicio parcial, con y sin generalización secundaria, en adultos y pacientes pediátricos de 3 años o más con epilepsia
DOSIFICACIÓN Y ADMINISTRACIÓN
Dosis para la neuralgia posherpética
En adultos con neuralgia posherpética, NEURONTIN puede iniciarse el Día 1 como una dosis única de 300 mg, el Día 2 como 600 mg/día (300 mg dos veces al día) y el Día 3 como 900 mg/día (300 mg tres veces al día). veces al día). Posteriormente, la dosis puede aumentarse según sea necesario para el alivio del dolor a una dosis de 1800 mg/día (600 mg tres veces al día). En estudios clínicos, se demostró la eficacia en un rango de dosis de 1800 mg/día a 3600 mg/día con efectos comparables en todo el rango de dosis; sin embargo, en estos estudios clínicos no se demostró el beneficio adicional de utilizar dosis superiores a 1800 mg/día.
Dosis para la epilepsia con convulsiones de inicio parcial
Pacientes de 12 años de edad y mayores
La dosis inicial es de 300 mg tres veces al día. La dosis de mantenimiento recomendada de NEURONTIN es de 300 mg a 600 mg tres veces al día. Dosis de hasta 2400 mg/día han sido bien toleradas en estudios clínicos a largo plazo. También se han administrado dosis de 3600 mg/día a un pequeño número de pacientes durante un tiempo relativamente corto y han sido bien toleradas. Administre NEURONTIN tres veces al día usando cápsulas de 300 mg o 400 mg, o tabletas de 600 mg u 800 mg. El tiempo máximo entre dosis no debe exceder las 12 horas.
Pacientes pediátricos de 3 a 11 años de edad
El rango de dosis inicial es de 10 mg/kg/día a 15 mg/kg/día, administrados en tres dosis divididas, y la dosis de mantenimiento recomendada se alcanza mediante una titulación ascendente durante un período de aproximadamente 3 días. La dosis de mantenimiento recomendada de NEURONTIN en pacientes de 3 a 4 años de edad es de 40 mg/kg/día, dividida en tres tomas. La dosis de mantenimiento recomendada de NEURONTIN 100 mg en pacientes de 5 a 11 años de edad es de 25 mg/kg/día a 35 mg/kg/día, administrados en tres dosis divididas. NEURONTIN 300 mg se puede administrar como solución oral, cápsula o tableta, o usando combinaciones de estas formulaciones. Dosis de hasta 50 mg/kg/día han sido bien toleradas en un estudio clínico a largo plazo. El intervalo de tiempo máximo entre dosis no debe exceder las 12 horas.
Ajuste de dosis en pacientes con insuficiencia renal
Se recomienda ajustar la dosis en pacientes de 12 años de edad y mayores con insuficiencia renal o sometidos a hemodiálisis, de la siguiente manera (consulte las recomendaciones de dosificación anteriores para conocer las dosis efectivas en cada indicación):
TABLA 1: Dosis de NEURONTIN basada en la función renal
El aclaramiento de creatinina (CLCr) es difícil de medir en pacientes ambulatorios. En pacientes con función renal estable, la depuración de creatinina puede estimarse razonablemente bien utilizando la ecuación de Cockcroft y Gault:
No se ha estudiado el uso de NEURONTIN en pacientes menores de 12 años con función renal comprometida.
Dosis en ancianos
Debido a que es más probable que los pacientes de edad avanzada tengan una función renal disminuida, se debe tener cuidado al seleccionar la dosis, y la dosis debe ajustarse en función de los valores de depuración de creatinina en estos pacientes.
Información de administración
Administrar NEURONTIN por vía oral con o sin alimentos.
Las cápsulas de NEURONTIN 100 mg deben tragarse enteras con agua.
Informe a los pacientes que, si dividen el comprimido ranurado de NEURONTIN de 600 mg u 800 mg para administrar medio comprimido, deben tomar el medio comprimido no utilizado como la siguiente dosis. Las medias tabletas que no se usen dentro de los 28 días posteriores a la división de la tableta ranurada deben desecharse.
Si la dosis de NEURONTIN se reduce, suspende o sustituye con un medicamento alternativo, esto debe hacerse gradualmente durante un mínimo de 1 semana (puede ser necesario un período más largo a discreción del prescriptor).
CÓMO SUMINISTRADO
Formas de dosificación y concentraciones
Cápsulas
- 100 mg: cápsulas de gelatina dura de color blanco impresas con “PD” en el cuerpo y “Neurontin/100 mg” en la tapa
- 300 mg: cápsulas de gelatina dura de color amarillo impresas con “PD” en el cuerpo y “Neurontin/300 mg” en la tapa
- 400 mg: cápsulas de gelatina dura de color naranja impresas con “PD” en el cuerpo y “Neurontin/400 mg” en la tapa
tabletas
- 600 mg: comprimidos ranurados recubiertos con película, elípticos, blancos, grabados con “NT” y “16” en una cara
- 800 mg: comprimidos recubiertos con película, elípticos, blancos, grabados con “NT” y “26” en una cara
Solucion Oral
- 250 mg por 5 ml (50 mg por ml), solución transparente incolora a ligeramente amarilla
Almacenamiento y manipulación
NEURONTIN (gabapentina) Las cápsulas, tabletas y solución oral se suministran de la siguiente manera:
Cápsulas de 100mg
Cápsulas de gelatina dura de color blanco impresas con “PD” en el cuerpo y “Neurontin/100 mg” en la tapa; disponible en:
Botellas de 100: CDN 0071-0803-24
Cápsulas de 300mg
Cápsulas de gelatina dura de color amarillo impresas con “PD” en el cuerpo y “Neurontin/300 mg” en la tapa; disponible en:
Botellas de 100: CDN 0071-0805-24 Dosis unitaria 50's: CDN 0071-0805-40
Cápsulas de 400mg
Cápsulas de gelatina dura de color naranja impresas con “PD” en el cuerpo y “Neurontin/400 mg” en la tapa; disponible en:
Botellas de 100: CDN 0071-0806-24 Dosis unitaria 50's: CDN 0071-0806-40
Comprimidos de 600mg
Comprimidos recubiertos con película, elípticos, de color blanco, grabados con “NT” y “16” en una cara; disponible en:
Botellas de 100: CDN 0071-0513-24
Comprimidos de 800 mg
Comprimidos recubiertos con película, elípticos, de color blanco, grabados con “NT” y “26” en una cara; disponible en:
Botellas de 100: CDN 0071-0401-24
250 mg por 5 ml de solución oral
Solución transparente de incolora a ligeramente amarilla; cada 5 ml de solución oral contiene 250 mg de gabapentina; disponible en:
Frascos de vidrio de 470 mL: CDN 0071-2012-23 Frascos de 470 mL: CDN 0071-2012-44
Guarde las tabletas y cápsulas NEURONTIN de 400 mg a 25 °C (77 °F); excursiones permitidas entre 15°C a 30°C (59°F a 86°F) [ver Temperatura ambiente controlada por USP ].
Guarde la solución oral de NEURONTIN refrigerada, entre 2 °C y 8 °C (entre 36 °F y 46 °F).
Distribuido por: Parke-Davis, División de Pfizer Inc., NY, NY 10017. Revisado: diciembre de 2020
EFECTOS SECUNDARIOS
Las siguientes reacciones adversas graves se analizan con mayor detalle en otras secciones:
- Reacción a medicamentos con eosinofilia y síntomas sistémicos (DRESS)/hipersensibilidad multiorgánica [ver ADVERTENCIAS Y PRECAUCIONES ]
- Anafilaxia y angioedema [ver ADVERTENCIAS Y PRECAUCIONES ]
- Somnolencia/Sedación y Mareos [ver ADVERTENCIAS Y PRECAUCIONES ]
- Convulsiones precipitadas por abstinencia, estado epiléptico [ver ADVERTENCIAS Y PRECAUCIONES ]
- Comportamiento e ideación suicida [ver ADVERTENCIAS Y PRECAUCIONES ]
- Depresión respiratoria [ver ADVERTENCIAS Y PRECAUCIONES ]
- Reacciones adversas neuropsiquiátricas (pacientes pediátricos de 3 a 12 años de edad) [ver ADVERTENCIAS Y PRECAUCIONES ]
- Muerte súbita e inexplicable en pacientes con epilepsia [ver ADVERTENCIAS Y PRECAUCIONES ]
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica.
Neuralgia postherpética
Las reacciones adversas más comunes asociadas con el uso de NEURONTIN en adultos, que no se observaron con una frecuencia equivalente entre los pacientes tratados con placebo, fueron mareos, somnolencia y edema periférico.
En los 2 ensayos controlados en neuralgia posherpética, el 16 % de los 336 pacientes que recibieron NEURONTIN y el 9 % de los 227 pacientes que recibieron placebo interrumpieron el tratamiento debido a una reacción adversa. Las reacciones adversas que más frecuentemente provocaron la abstinencia en los pacientes tratados con NEURONTIN fueron mareos, somnolencia y náuseas.
La Tabla 3 enumera las reacciones adversas que ocurrieron en al menos el 1 % de los pacientes con neuralgia posherpética tratados con NEURONTIN que participaron en ensayos controlados con placebo y que fueron numéricamente más frecuentes en el grupo de NEURONTIN que en el grupo de placebo.
TABLA 3: Reacciones adversas en ensayos combinados controlados con placebo en neuralgia posherpética
Otras reacciones en más del 1% de los pacientes, pero igualmente o más frecuentes en el grupo placebo, incluyeron dolor, temblor, neuralgia, dolor de espalda, dispepsia, disnea y síndrome gripal.
No hubo diferencias clínicamente importantes entre hombres y mujeres en los tipos e incidencia de reacciones adversas. Debido a que hubo pocos pacientes cuya raza se informó como diferente a la blanca, no hay datos suficientes para respaldar una declaración sobre la distribución de reacciones adversas por raza.
Epilepsia con convulsiones de inicio parcial (terapia complementaria)
Las reacciones adversas más frecuentes con NEURONTIN 300 mg en combinación con otros fármacos antiepilépticos en pacientes mayores de 12 años, que no se observaron con una frecuencia equivalente entre los pacientes tratados con placebo, fueron somnolencia, mareos, ataxia, fatiga y nistagmo.
Las reacciones adversas más comunes con NEURONTIN en combinación con otros medicamentos antiepilépticos en pacientes pediátricos de 3 a 12 años de edad, que no se observaron con la misma frecuencia entre los pacientes tratados con placebo, fueron infección viral, fiebre, náuseas y/o vómitos, somnolencia y hostilidad [ver ADVERTENCIAS Y PRECAUCIONES ].
Aproximadamente el 7 % de los 2074 pacientes mayores de 12 años y aproximadamente el 7 % de los 449 pacientes pediátricos de 3 a 12 años que recibieron NEURONTIN en los ensayos clínicos previos a la comercialización interrumpieron el tratamiento debido a una reacción adversa. Las reacciones adversas más comúnmente asociadas con la abstinencia en pacientes mayores de 12 años fueron somnolencia (1,2 %), ataxia (0,8 %), fatiga (0,6 %), náuseas y/o vómitos (0,6 %) y mareos (0,6 %). . Las reacciones adversas más comúnmente asociadas con la abstinencia en pacientes pediátricos fueron labilidad emocional (1,6 %), hostilidad (1,3 %) e hipercinesia (1,1 %).
La Tabla 4 enumera las reacciones adversas que ocurrieron en al menos el 1 % de los pacientes mayores de 12 años con epilepsia tratados con NEURONTIN que participaron en ensayos controlados con placebo y fueron numéricamente más comunes en el grupo de NEURONTIN. En estos estudios, se agregó NEURONTIN 300 mg o placebo a la terapia actual con medicamentos antiepilépticos del paciente.
TABLA 4: Reacciones adversas en ensayos complementarios combinados controlados con placebo en pacientes con epilepsia >12 años de edad
Entre las reacciones adversas que se produjeron con una incidencia de al menos el 10 % en los pacientes tratados con NEURONTIN, la somnolencia y la ataxia parecieron mostrar una relación dosis-respuesta positiva.
La incidencia general de reacciones adversas y los tipos de reacciones adversas observados fueron similares entre hombres y mujeres tratados con NEURONTIN. La incidencia de reacciones adversas aumentó ligeramente con el aumento de la edad en pacientes tratados con NEURONTIN o con placebo. Debido a que solo el 3% de los pacientes (28/921) en los estudios controlados con placebo se identificaron como no blancos (negros u otros), no hay datos suficientes para respaldar una declaración sobre la distribución de las reacciones adversas por raza.
La Tabla 5 enumera las reacciones adversas que ocurrieron en al menos el 2% de los pacientes tratados con NEURONTIN, de 3 a 12 años de edad con epilepsia que participaron en ensayos controlados con placebo, y que fueron numéricamente más comunes en el grupo de NEURONTIN.
TABLA 5: Reacciones adversas en un ensayo adicional controlado con placebo en pacientes pediátricos con epilepsia de 3 a 12 años de edad
Otras reacciones en más del 2% de los pacientes pediátricos de 3 a 12 años de edad, pero con igual o mayor frecuencia en el grupo de placebo incluyeron: faringitis, infección de las vías respiratorias superiores, dolor de cabeza, rinitis, convulsiones, diarrea, anorexia, tos y otitis media.
Experiencia posterior a la comercialización
Se han identificado las siguientes reacciones adversas durante el uso posterior a la comercialización de NEURONTIN. Debido a que estas reacciones son informadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Trastornos hepatobiliares: ictericia
Investigaciones: creatina quinasa elevada, pruebas de función hepática elevadas
Trastornos del metabolismo y de la nutrición: hiponatremia
Trastorno musculoesquelético y del tejido conjuntivo: rabdomiolisis
Trastornos del sistema nervioso: trastorno del movimiento
Desórdenes psiquiátricos: agitación
Trastornos del aparato reproductor y de la mama: agrandamiento de los senos, cambios en la libido, trastornos de la eyaculación y anorgasmia
Trastornos de la piel y del tejido subcutáneo: angioedema [ver ADVERTENCIAS Y PRECAUCIONES ], penfigoide ampolloso, eritema multiforme, síndrome de Stevens-Johnson.
Hay informes posteriores a la comercialización de depresión respiratoria mortal o potencialmente mortal en pacientes que toman NEURONTIN con opioides u otros depresores del SNC, o en el contexto de una insuficiencia respiratoria subyacente [ver ADVERTENCIAS Y PRECAUCIONES ].
También se han notificado reacciones adversas tras la interrupción brusca de la gabapentina. Las reacciones informadas con mayor frecuencia fueron ansiedad, insomnio, náuseas, dolor y sudoración.
INTERACCIONES CON LA DROGAS
opioides
Se han notificado depresión respiratoria y sedación, que a veces provocan la muerte, después de la coadministración de gabapentina con opioides (p. ej., morfina, hidrocodona, oxicodona, buprenorfina) [ver ADVERTENCIAS Y PRECAUCIONES ].
hidrocodona
La coadministración de NEURONTIN 300 mg con hidrocodona disminuye la exposición a hidrocodona [ver FARMACOLOGÍA CLÍNICA ]. Se debe considerar el potencial de alteración en la exposición y el efecto de hidrocodona cuando se inicia o interrumpe NEURONTIN 100 mg en un paciente que toma hidrocodona.
Morfina
Cuando se administra gabapentina con morfina, se debe observar a los pacientes para detectar signos de depresión del SNC, como somnolencia, sedación y depresión respiratoria [ver FARMACOLOGÍA CLÍNICA ].
Otros medicamentos antiepilépticos
La gabapentina no se metaboliza de forma apreciable ni interfiere con el metabolismo de los fármacos antiepilépticos comúnmente coadministrados [ver FARMACOLOGÍA CLÍNICA ].
Maalox® (hidróxido de aluminio, hidróxido de magnesio)
La biodisponibilidad media de la gabapentina se redujo en aproximadamente un 20 % con el uso concomitante de un antiácido (Maalox) que contiene hidróxidos de magnesio y aluminio. Se recomienda tomar gabapentina al menos 2 horas después de la administración de Maalox [ver FARMACOLOGÍA CLÍNICA ].
Interacciones entre medicamentos y pruebas de laboratorio
Debido a que se informaron lecturas falsas positivas con la prueba de tira reactiva Ames N-Multistix SG para proteína urinaria cuando se agregó gabapentina a otros medicamentos antiepilépticos, se recomienda el procedimiento de precipitación con ácido sulfosalicílico más específico para determinar la presencia de proteína en orina.
Abuso y dependencia de drogas
Sustancia controlada
La gabapentina no es un fármaco catalogado.
Abuso
El abuso es el uso intencional y no terapéutico de una droga, aunque sea una sola vez, por sus efectos psicológicos o fisiológicos deseables. El uso indebido es el uso intencional, con fines terapéuticos, de un medicamento por parte de un individuo de una manera distinta a la prescrita por un proveedor de atención médica o para quien no fue prescrito.
La gabapentina no muestra afinidad por los sitios receptores de benzodiacepinas, opioides (mu, delta o kappa) o cannabinoides 1. El uso indebido y el abuso de gabapentina se han informado en el entorno posterior a la comercialización y en la literatura publicada. La mayoría de las personas descritas en estos informes tenían antecedentes de abuso de varias sustancias. Algunas de estas personas estaban tomando dosis de gabapentina superiores a las recomendadas para usos no aprobados. Al prescribir NEURONTIN, evalúe cuidadosamente a los pacientes en busca de antecedentes de abuso de drogas y obsérvelos para detectar signos y síntomas de uso indebido o abuso de gabapentina (p. ej., aumento de la autodosis y comportamiento de búsqueda de drogas). El potencial de abuso de la gabapentina no ha sido evaluado en estudios en humanos.
Dependencia
La dependencia física es un estado que se desarrolla como resultado de la adaptación fisiológica en respuesta al uso repetido de drogas, que se manifiesta por signos y síntomas de abstinencia después de una interrupción abrupta o una reducción significativa de la dosis de una droga. Hay informes posteriores a la comercialización poco comunes de personas que experimentan síntomas de abstinencia poco después de suspender las dosis de gabapentina más altas que las recomendadas para tratar enfermedades para las cuales el medicamento no está aprobado. Dichos síntomas incluyeron agitación, desorientación y confusión después de suspender repentinamente la gabapentina que se resolvió después de reiniciar la gabapentina. El potencial de dependencia de la gabapentina no ha sido evaluado en estudios en humanos.
ADVERTENCIAS
Incluido como parte de la PRECAUCIONES sección.
PRECAUCIONES
Reacción a medicamentos con eosinofilia y síntomas sistémicos (DRESS)/hipersensibilidad multiorgánica
Se ha producido una reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS), también conocida como hipersensibilidad multiorgánica, con NEURONTIN. Algunas de estas reacciones han sido fatales o potencialmente mortales. DRESS típicamente, aunque no exclusivamente, se presenta con fiebre, erupción cutánea y/o linfadenopatía, en asociación con otros compromisos de órganos, como hepatitis, nefritis, anomalías hematológicas, miocarditis o miositis que a veces se asemejan a una infección viral aguda. La eosinofilia a menudo está presente. Este trastorno es variable en su expresión y pueden estar involucrados otros sistemas de órganos que no se mencionan aquí.
Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad, como fiebre o linfadenopatía, pueden estar presentes aunque la erupción no sea evidente. Si tales signos o síntomas están presentes, el paciente debe ser evaluado inmediatamente. NEURONTIN debe suspenderse si no se puede establecer una etiología alternativa para los signos o síntomas.
Anafilaxia y angioedema
NEURONTIN puede causar anafilaxia y angioedema después de la primera dosis o en cualquier momento durante el tratamiento. Los signos y síntomas en los casos informados incluyen dificultad para respirar, hinchazón de los labios, la garganta y la lengua, e hipotensión que requiere tratamiento de emergencia. Se debe indicar a los pacientes que suspendan NEURONTIN 300 mg y busquen atención médica inmediata si experimentan signos o síntomas de anafilaxia o angioedema.
Efectos sobre la conducción y operación de maquinaria pesada
Los pacientes que toman NEURONTIN no deben conducir hasta que hayan adquirido suficiente experiencia para evaluar si NEURONTIN 100 mg afecta su capacidad para conducir. Los estudios de rendimiento de conducción realizados con un profármaco de gabapentina (tableta de gabapentina enacarbil, de liberación prolongada) indican que la gabapentina puede causar un deterioro significativo de la conducción. Los prescriptores y los pacientes deben ser conscientes de que los pacientes' la capacidad para evaluar su propia capacidad de conducción, así como su capacidad para evaluar el grado de somnolencia causado por NEURONTIN, puede ser imperfecta. Se desconoce la duración de la dificultad para conducir después de iniciar la terapia con NEURONTIN 300 mg. Si el deterioro está relacionado con la somnolencia [ver ADVERTENCIAS Y PRECAUCIONES ] u otros efectos de NEURONTIN son desconocidos.
Además, dado que NEURONTIN 300 mg provoca somnolencia y mareos [ver ADVERTENCIAS Y PRECAUCIONES ], se debe advertir a los pacientes que no operen maquinaria compleja hasta que hayan adquirido suficiente experiencia con NEURONTIN para evaluar si NEURONTIN 300 mg afecta su capacidad para realizar dichas tareas.
Somnolencia/Sedación Y Mareos
Durante los ensayos controlados de epilepsia en pacientes mayores de 12 años que recibieron dosis de NEURONTIN de 100 mg a 1800 mg al día, se informó una tasa mayor de somnolencia, mareos y ataxia en pacientes que recibieron NEURONTIN en comparación con placebo: es decir, 19 % en pacientes que recibieron el fármaco. frente al 9 % con placebo para la somnolencia, el 17 % con el fármaco frente al 7 % con placebo para los mareos y el 13 % con el fármaco frente al 6 % con placebo para la ataxia. En estos ensayos, la somnolencia, la ataxia y la fatiga fueron reacciones adversas frecuentes que llevaron a la suspensión de NEURONTIN en pacientes mayores de 12 años, con un 1,2 %, 0,8 % y 0,6 % que interrumpieron por estos eventos, respectivamente.
Durante los ensayos controlados en pacientes con neuralgia posherpética, la somnolencia y los mareos se informaron a un ritmo mayor en comparación con el placebo en pacientes que recibieron NEURONTIN, en dosis de hasta 3600 mg por día: es decir, 21 % en pacientes tratados con NEURONTIN versus 5 % en pacientes tratados con placebo para somnolencia y 28 % en pacientes tratados con NEURONTIN versus 8 % en pacientes tratados con placebo para mareos. Los mareos y la somnolencia se encontraban entre las reacciones adversas más comunes que llevaron a la suspensión de NEURONTIN.
Se debe observar cuidadosamente a los pacientes en busca de signos de depresión del sistema nervioso central (SNC), como somnolencia y sedación, cuando NEURONTIN 400 mg se usa con otros medicamentos con propiedades sedantes debido a la sinergia potencial. Además, los pacientes que requieren tratamiento concomitante con morfina pueden experimentar aumentos en las concentraciones de gabapentina y pueden requerir un ajuste de dosis [ver INTERACCIONES CON LA DROGAS ].
Convulsión precipitada por abstinencia, estado epiléptico
Los medicamentos antiepilépticos no deben suspenderse abruptamente debido a la posibilidad de aumentar la frecuencia de las convulsiones.
En los estudios de epilepsia controlados con placebo en pacientes mayores de 12 años, la incidencia de estado epiléptico en pacientes que recibieron NEURONTIN fue del 0,6 % (3 de 543) frente al 0,5 % en pacientes que recibieron placebo (2 de 378). Entre los 2074 pacientes mayores de 12 años tratados con NEURONTIN 300 mg en todos los estudios de epilepsia (controlados y no controlados), 31 (1,5 %) tenían estado epiléptico. De estos, 14 pacientes no tenían antecedentes de estado epiléptico antes del tratamiento o mientras tomaban otros medicamentos. Debido a que no se dispone de datos históricos adecuados, es imposible decir si el tratamiento con NEURONTIN se asocia o no con una tasa de estado epiléptico más alta o más baja de lo que se esperaría que ocurriera en una población similar no tratada con NEURONTIN.
Comportamiento e ideación suicida
Los fármacos antiepilépticos (FAE), incluido NEURONTIN 100 mg, aumentan el riesgo de pensamientos o conductas suicidas en pacientes que toman estos fármacos por cualquier indicación. Los pacientes tratados con cualquier AED por cualquier indicación deben ser monitoreados para detectar la aparición o el empeoramiento de depresión, pensamientos o comportamientos suicidas y/o cualquier cambio inusual en el estado de ánimo o el comportamiento.
Los análisis agrupados de 199 ensayos clínicos controlados con placebo (terapia mono y adyuvante) de 11 FAE diferentes mostraron que los pacientes asignados al azar a uno de los FAE tenían aproximadamente el doble de riesgo (riesgo relativo ajustado 1,8, IC del 95%: 1,2, 2,7) de suicidio. pensamiento o comportamiento en comparación con los pacientes asignados al azar a placebo. En estos ensayos, que tuvieron una mediana de duración del tratamiento de 12 semanas, la tasa de incidencia estimada de ideación o conducta suicida entre 27 863 pacientes tratados con AED fue del 0,43 %, en comparación con el 0,24 % entre 16 029 pacientes tratados con placebo, lo que representa un aumento de aproximadamente un caso de pensamiento o conducta suicida por cada 530 pacientes atendidos. Hubo cuatro suicidios en pacientes tratados con fármacos en los ensayos y ninguno en pacientes tratados con placebo, pero el número es demasiado pequeño para permitir una conclusión sobre el efecto del fármaco en el suicidio.
El aumento del riesgo de pensamientos o conductas suicidas con los FAE se observó tan pronto como una semana después de comenzar el tratamiento farmacológico con FAE y persistió durante la duración del tratamiento evaluado. Debido a que la mayoría de los ensayos incluidos en el análisis no se extendieron más allá de las 24 semanas, no se pudo evaluar el riesgo de pensamientos o conductas suicidas más allá de las 24 semanas.
El riesgo de pensamientos o conductas suicidas fue generalmente consistente entre las drogas en los datos analizados. El hallazgo de un mayor riesgo con los AED de diversos mecanismos de acción y en una variedad de indicaciones sugiere que el riesgo se aplica a todos los AED utilizados para cualquier indicación. El riesgo no varió sustancialmente según la edad (5-100 años) en los ensayos clínicos analizados. La Tabla 2 muestra el riesgo absoluto y relativo por indicación para todos los FAE evaluados.
TABLA 2: Riesgo por indicación de fármacos antiepilépticos en el análisis combinado
El riesgo relativo de pensamientos o conductas suicidas fue mayor en los ensayos clínicos de epilepsia que en los ensayos clínicos de enfermedades psiquiátricas u otras, pero las diferencias de riesgo absoluto fueron similares para las indicaciones de epilepsia y psiquiátricas.
Cualquiera que esté considerando recetar NEURONTIN 100 mg o cualquier otro AED debe sopesar el riesgo de pensamientos o conductas suicidas con el riesgo de una enfermedad no tratada. La epilepsia y muchas otras enfermedades para las que se recetan FAE están asociadas con morbilidad y mortalidad y un mayor riesgo de pensamientos y conductas suicidas. Si surgen pensamientos y conductas suicidas durante el tratamiento, el prescriptor debe considerar si la aparición de estos síntomas en un paciente determinado puede estar relacionada con la enfermedad que se está tratando.
Se debe informar a los pacientes, sus cuidadores y familias que los AED aumentan el riesgo de pensamientos y comportamientos suicidas y se les debe advertir sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los signos y síntomas de depresión, cualquier cambio inusual en el estado de ánimo o el comportamiento. , o la aparición de pensamientos o conductas suicidas o pensamientos de autolesión. Los comportamientos de preocupación deben informarse de inmediato a los proveedores de atención médica.
Depresion respiratoria
Existe evidencia de informes de casos, estudios en humanos y estudios en animales que asocian la gabapentina con depresión respiratoria grave, potencialmente mortal o fatal cuando se administra junto con depresores del SNC, incluidos los opioides, o en el contexto de una insuficiencia respiratoria subyacente. Cuando se toma la decisión de recetar conjuntamente NEURONTIN 100 mg con otro depresor del SNC, en particular un opioide, o recetar NEURONTIN 100 mg a pacientes con insuficiencia respiratoria subyacente, controle a los pacientes para detectar síntomas de depresión respiratoria y sedación, y considere iniciar NEURONTIN a una dosis baja. . El tratamiento de la depresión respiratoria puede incluir una estrecha observación, medidas de apoyo y reducción o retirada de los depresores del SNC (incluido NEURONTIN).
Reacciones adversas neuropsiquiátricas (pacientes pediátricos de 3 a 12 años de edad)
El uso de gabapentina en pacientes pediátricos con epilepsia de 3 a 12 años de edad está asociado con la aparición de reacciones adversas relacionadas con el SNC. Los más significativos se pueden clasificar en las siguientes categorías: 1) labilidad emocional (principalmente problemas de comportamiento), 2) hostilidad, incluidos los comportamientos agresivos, 3) trastorno del pensamiento, incluidos los problemas de concentración y cambios en el rendimiento escolar, y 4) hipercinesia ( principalmente inquietud e hiperactividad). Entre los pacientes tratados con gabapentina, la mayoría de las reacciones fueron de intensidad leve a moderada.
En ensayos clínicos controlados de epilepsia en pacientes pediátricos de 3 a 12 años de edad, la incidencia de estas reacciones adversas fue: labilidad emocional 6 % (pacientes tratados con gabapentina) versus 1,3 % (pacientes tratados con placebo); hostilidad 5,2% versus 1,3%; hipercinesia 4,7% versus 2,9%; y trastorno del pensamiento 1,7% versus 0%. Una de estas reacciones, un informe de hostilidad, se consideró grave. La interrupción del tratamiento con gabapentina se produjo en el 1,3 % de los pacientes que informaron labilidad emocional e hipercinesia y en el 0,9 % de los pacientes tratados con gabapentina que informaron hostilidad y trastornos del pensamiento. Un paciente tratado con placebo (0,4 %) se retiró debido a labilidad emocional.
Potencial tumorigénico
En un estudio de carcinogenicidad oral, la gabapentina aumentó la incidencia de tumores de células acinares pancreáticas en ratas [ver Toxicología no clínica ]. La importancia clínica de este hallazgo es desconocida. La experiencia clínica durante el desarrollo previo a la comercialización de la gabapentina no proporciona medios directos para evaluar su potencial para inducir tumores en humanos.
En estudios clínicos de terapia adyuvante en epilepsia que incluyeron 2085 años-paciente de exposición en pacientes >12 años de edad, se informaron nuevos tumores en 10 pacientes (2 de mama, 3 de cerebro, 2 de pulmón, 1 suprarrenal, 1 no Hodgkin' s, 1 carcinoma endometrial in situ) y los tumores preexistentes empeoraron en 11 pacientes (9 de cerebro, 1 de mama, 1 de próstata) durante o hasta 2 años después de la suspensión de NEURONTIN. Sin el conocimiento de la incidencia de fondo y la recurrencia en una población similar no tratada con NEURONTIN 100 mg, es imposible saber si la incidencia observada en esta cohorte se ve afectada o no por el tratamiento.
Muerte súbita e inexplicable en pacientes con epilepsia
Durante el curso del desarrollo previo a la comercialización de NEURONTIN 400 mg, se registraron 8 muertes súbitas e inexplicables entre una cohorte de 2203 pacientes con epilepsia tratados (2103 años-paciente de exposición) con NEURONTIN.
Algunos de estos podrían representar muertes relacionadas con convulsiones en las que no se observó la convulsión, por ejemplo, durante la noche. Esto representa una incidencia de 0,0038 muertes por paciente-año. Aunque esta tasa supera la esperada en una población sana emparejada por edad y sexo, se encuentra dentro del rango de estimaciones de la incidencia de muertes súbitas inexplicables en pacientes con epilepsia que no reciben NEURONTIN (que van desde 0,0005 para la población general de epilépticos hasta 0,003 para una población de ensayo clínico similar a la del programa NEURONTIN, a 0,005 para pacientes con epilepsia refractaria). En consecuencia, si estas cifras son tranquilizadoras o generan más preocupación depende de la comparabilidad de las poblaciones informadas a la cohorte NEURONTIN y la precisión de las estimaciones proporcionadas.
Información de asesoramiento para pacientes
Aconseje al paciente que lea la etiqueta del paciente aprobada por la FDA ( Guía de medicamentos ).
Información de administración
Informe a los pacientes que NEURONTIN se toma por vía oral con o sin alimentos. Informe a los pacientes que, si dividen la tableta ranurada de 600 mg u 800 mg para administrar la mitad de la tableta, deben tomar la mitad de la tableta no utilizada como la siguiente dosis. Aconseje a los pacientes que desechen las medias tabletas que no hayan usado dentro de los 28 días posteriores a la división de la tableta ranurada.
Reacción a medicamentos con eosinofilia y síntomas sistémicos (DRESS)/hipersensibilidad multiorgánica
Antes de iniciar el tratamiento con NEURONTIN, instruya a los pacientes que una erupción u otros signos o síntomas de hipersensibilidad (como fiebre o linfadenopatía) pueden anunciar un evento médico grave y que el paciente debe informar inmediatamente a un médico sobre cualquier evento de este tipo [ver ADVERTENCIAS Y PRECAUCIONES ].
Anafilaxia y angioedema
Aconseje a los pacientes que descontinúen NEURONTIN 300 mg y busquen atención médica si desarrollan signos o síntomas de anafilaxia o angioedema [ver ADVERTENCIAS Y PRECAUCIONES ].
Mareos y somnolencia y efectos sobre la conducción y operación de maquinaria pesada
Advierta a los pacientes que NEURONTIN 100 mg puede causar mareos, somnolencia y otros síntomas y signos de depresión del SNC. Otros medicamentos con propiedades sedantes pueden aumentar estos síntomas. En consecuencia, aunque los pacientes' capacidad para determinar su nivel de discapacidad puede ser poco fiable, aconséjeles que no conduzcan un coche ni operen otra maquinaria compleja hasta que hayan adquirido suficiente experiencia con NEURONTIN para evaluar si afecta negativamente a su rendimiento mental y/o motor. Informe a los pacientes que no se sabe cuánto tiempo dura este efecto [ver ADVERTENCIAS Y PRECAUCIONES y ADVERTENCIAS Y PRECAUCIONES ].
Pensamiento y comportamiento suicida
Aconseje al paciente, a sus cuidadores y a sus familias que los DEA, incluido NEURONTIN, pueden aumentar el riesgo de pensamientos y conductas suicidas. Advierta a los pacientes sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los síntomas de depresión, cualquier cambio inusual en el estado de ánimo o el comportamiento, o la aparición de pensamientos, conductas o pensamientos suicidas sobre autolesión. Indique a los pacientes que informen inmediatamente a los proveedores de atención médica sobre los comportamientos que les preocupan [ver ADVERTENCIAS Y PRECAUCIONES ].
Depresion respiratoria
Informar a los pacientes sobre el riesgo de depresión respiratoria. Incluya información de que el riesgo es mayor para aquellos que usan depresores del SNC concomitantes (como analgésicos opioides) o aquellos con insuficiencia respiratoria subyacente. Enseñe a los pacientes cómo reconocer la depresión respiratoria y aconséjeles que busquen atención médica de inmediato si ocurre [ver ADVERTENCIAS Y PRECAUCIONES ].
Uso en el embarazo
Indique a los pacientes que notifiquen a su médico si quedan embarazadas o tienen la intención de quedar embarazadas durante la terapia, y que notifiquen a su médico si están amamantando o tienen la intención de amamantar durante la terapia [ver Uso en poblaciones específicas ].
Aliente a las pacientes a inscribirse en el Registro de embarazos de la NAAED si quedan embarazadas. Este registro recopila información sobre la seguridad de los medicamentos antiepilépticos durante el embarazo. Para inscribirse, los pacientes pueden llamar al número gratuito 1-888-233-2334 [ver Uso en poblaciones específicas ].
Es posible que se haya actualizado el etiquetado de este producto. Para obtener la información de prescripción más reciente, visite www.pfizer.com.
Toxicología no clínica
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
Carcinogénesis
La gabapentina se administró por vía oral a ratones y ratas en estudios de carcinogenicidad de 2 años. No se observó evidencia de carcinogenicidad relacionada con el fármaco en ratones tratados con dosis de hasta 2000 mg/kg/día. A 2000 mg/kg, la exposición plasmática a gabapentina (AUC) en ratones fue aproximadamente 2 veces mayor que en humanos a la MRHD de 3600 mg/día. En ratas, se encontraron aumentos en la incidencia de adenoma y carcinoma de células acinares pancreáticas en ratas macho que recibieron la dosis más alta (2000 mg/kg), pero no en dosis de 250 o 1000 mg/kg/día. A 1000 mg/kg, la exposición a la gabapentina en plasma (AUC) en ratas fue aproximadamente 5 veces mayor que la de los seres humanos en la MRHD.
Los estudios diseñados para investigar el mecanismo de carcinogénesis pancreática inducida por gabapentina en ratas indican que la gabapentina estimula la síntesis de ADN en células acinares pancreáticas de rata in vitro y, por lo tanto, puede actuar como un promotor tumoral al aumentar la actividad mitogénica. No se sabe si la gabapentina tiene la capacidad de aumentar la proliferación celular en otros tipos de células o en otras especies, incluidos los humanos.
mutagénesis
La gabapentina no demostró potencial mutagénico o genotóxico in vitro (prueba de Ames, ensayo de mutación directa HGPRT en células de pulmón de hámster chino) e in vivo (prueba de aberración cromosómica y micronúcleo en médula ósea de hámster chino, micronúcleo de ratón, síntesis de ADN no programada en hepatocitos de rata) ensayos
Deterioro de la fertilidad
No se observaron efectos adversos sobre la fertilidad o la reproducción en ratas a dosis de hasta 2000 mg/kg. A 2000 mg/kg, la exposición a la gabapentina en plasma (AUC) en ratas es aproximadamente 8 veces mayor que la de los seres humanos en la MRHD.
Uso en poblaciones específicas
El embarazo
Registro de exposición durante el embarazo
Existe un registro de exposición durante el embarazo que monitorea los resultados del embarazo en mujeres expuestas a medicamentos antiepilépticos (FAE), como NEURONTIN 300 mg, durante el embarazo. Anime a las mujeres que toman NEURONTIN durante el embarazo a inscribirse en el Registro de Embarazo de Medicamentos Antiepilépticos de América del Norte (NAAED) llamando al número gratuito 1-888-233-2334 o visitando http://www.aedpregnancyregistry.org/.
Resumen de riesgos
No hay datos adecuados sobre los riesgos de desarrollo asociados con el uso de NEURONTIN en mujeres embarazadas. En estudios no clínicos en ratones, ratas y conejos, la gabapentina fue tóxica para el desarrollo (aumentó las anomalías viscerales y esqueléticas fetales y aumentó la mortalidad embriofetal) cuando se administró a animales preñados en dosis similares o inferiores a las utilizadas clínicamente [ver Datos ].
En la población general de los EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 al 4 % y del 15 al 20 %, respectivamente. Se desconoce el riesgo de fondo de defectos congénitos importantes y abortos espontáneos para la población indicada.
Datos
Datos de animales
Cuando los ratones preñados recibieron dosis orales de gabapentina (500, 1000 o 3000 mg/kg/día) durante el período de organogénesis, se observó toxicidad embriofetal (mayor incidencia de variaciones esqueléticas) en las dos dosis más altas. La dosis sin efecto para la toxicidad del desarrollo embriofetal en ratones (500 mg/kg/día) es menor que la dosis máxima recomendada en humanos (MRHD) de 3600 mg sobre la base del área de superficie corporal (mg/m²).
En estudios en los que las ratas recibieron dosis orales de gabapentina (500 a 2000 mg/kg/día) durante el embarazo, se observaron efectos adversos en el desarrollo de las crías (aumento de la incidencia de hidrouréter y/o hidronefrosis) con todas las dosis. La dosis más baja probada es similar a la MRHD en mg/m².
Cuando se trató a conejas preñadas con gabapentina durante el período de organogénesis, se observó un aumento en la mortalidad embriofetal en todas las dosis probadas (60, 300 o 1500 mg/kg). La dosis más baja probada es menor que la MRHD en mg/m².
En un estudio publicado, se administró gabapentina (400 mg/kg/día) mediante inyección intraperitoneal a ratones recién nacidos durante la primera semana posnatal, un período de sinaptogénesis en roedores (que corresponde al último trimestre del embarazo en humanos). La gabapentina causó una marcada disminución en la formación de sinapsis neuronales en cerebros de ratones intactos y formación anormal de sinapsis neuronales en un modelo de ratón de reparación sináptica. Se ha demostrado in vitro que la gabapentina interfiere con la actividad de la subunidad α2δ de los canales de calcio activados por voltaje, un receptor involucrado en la sinaptogénesis neuronal. Se desconoce el significado clínico de estos hallazgos.
Lactancia
Resumen de riesgos
La gabapentina se secreta en la leche humana después de la administración oral. Se desconocen los efectos sobre el lactante y sobre la producción de leche. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de NEURONTIN de la madre y cualquier efecto adverso potencial en el lactante a causa de NEURONTIN o de la afección materna subyacente.
Uso pediátrico
No se ha establecido la seguridad y eficacia de NEURONTIN 600 mg en el tratamiento de la neuralgia posherpética en pacientes pediátricos.
No se ha establecido la seguridad y eficacia como terapia adyuvante en el tratamiento de convulsiones parciales en pacientes pediátricos menores de 3 años [ver Estudios clínicos ].
Uso geriátrico
El número total de pacientes tratados con NEURONTIN en ensayos clínicos controlados en pacientes con neuralgia posherpética fue de 336, de los cuales 102 (30 %) tenían entre 65 y 74 años y 168 (50 %) tenían 75 años o más. Hubo un mayor efecto del tratamiento en pacientes de 75 años de edad y mayores en comparación con pacientes más jóvenes que recibieron la misma dosis. Dado que la gabapentina se elimina casi exclusivamente por excreción renal, el mayor efecto del tratamiento observado en pacientes ≥75 años puede ser consecuencia de una mayor exposición a la gabapentina para una dosis dada que resulta de una disminución de la función renal relacionada con la edad. Sin embargo, no se pueden excluir otros factores. Los tipos y la incidencia de las reacciones adversas fueron similares en todos los grupos de edad, excepto por el edema periférico y la ataxia, cuya incidencia tendió a aumentar con la edad.
Los estudios clínicos de NEURONTIN en epilepsia no incluyeron un número suficiente de sujetos de 65 años o más para determinar si respondían de manera diferente a los sujetos más jóvenes. Otra experiencia clínica informada no ha identificado diferencias en las respuestas entre los ancianos y los pacientes más jóvenes. En general, la selección de la dosis para un paciente de edad avanzada debe ser cautelosa, generalmente comenzando en el extremo inferior del rango de dosificación, lo que refleja la mayor frecuencia de disminución de la función hepática, renal o cardíaca, y de enfermedades concomitantes u otra terapia con medicamentos.
Se sabe que este fármaco se excreta sustancialmente por vía renal, y el riesgo de reacciones tóxicas a este fármaco puede ser mayor en pacientes con insuficiencia renal. Debido a que es más probable que los pacientes de edad avanzada tengan una función renal disminuida, se debe tener cuidado al seleccionar la dosis, y la dosis debe ajustarse en función de los valores de depuración de creatinina en estos pacientes [ver DOSIFICACIÓN Y ADMINISTRACIÓN , REACCIONES ADVERSAS , y FARMACOLOGÍA CLÍNICA ].
Insuficiencia renal
Es necesario ajustar la dosis en pacientes adultos con función renal comprometida [ver DOSIFICACIÓN Y ADMINISTRACIÓN y FARMACOLOGÍA CLÍNICA ]. No se han estudiado pacientes pediátricos con insuficiencia renal.
Es necesario ajustar la dosis en pacientes sometidos a hemodiálisis [ver DOSIFICACIÓN Y ADMINISTRACIÓN y FARMACOLOGÍA CLÍNICA ].
SOBREDOSIS
Los signos de toxicidad aguda en animales incluyeron ataxia, dificultad para respirar, ptosis, sedación, hipoactividad o excitación.
Se han informado sobredosis orales agudas de NEURONTIN 300 mg. Los síntomas han incluido visión doble, temblor, dificultad para hablar, somnolencia, estado mental alterado, mareos, letargo y diarrea. Se ha informado depresión respiratoria fatal con sobredosis de 400 mg de NEURONTIN, solo y en combinación con otros depresores del SNC.
La gabapentina puede eliminarse mediante hemodiálisis.
Si ocurre una sobreexposición, llame a su centro de control de envenenamiento al 1-800-222-1222.
CONTRAINDICACIONES
NEURONTIN está contraindicado en pacientes que han demostrado hipersensibilidad al fármaco oa sus ingredientes.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
Se desconocen los mecanismos precisos por los cuales la gabapentina produce sus acciones analgésicas y antiepilépticas. La gabapentina está estructuralmente relacionada con el neurotransmisor ácido gamma-aminobutírico (GABA), pero no tiene efecto sobre la unión, captación o degradación de GABA. Los estudios in vitro han demostrado que la gabapentina se une con gran afinidad a la subunidad α2δ de los canales de calcio activados por voltaje; sin embargo, se desconoce la relación de esta unión con los efectos terapéuticos de la gabapentina.
Farmacocinética
Todas las acciones farmacológicas posteriores a la administración de gabapentina se deben a la actividad del compuesto original; la gabapentina no se metaboliza apreciablemente en humanos.
Biodisponibilidad Oral
La biodisponibilidad de la gabapentina no es proporcional a la dosis; es decir, a medida que aumenta la dosis, disminuye la biodisponibilidad. La biodisponibilidad de la gabapentina es de aproximadamente 60 %, 47 %, 34 %, 33 % y 27 % luego de 900, 1200, 2400, 3600 y 4800 mg/día administrados en 3 dosis divididas, respectivamente. Los alimentos tienen solo un ligero efecto sobre la velocidad y el grado de absorción de la gabapentina (aumento del 14 % en el AUC y la Cmax).
Distribución
Menos del 3% de la gabapentina circula unida a proteínas plasmáticas. El volumen de distribución aparente de la gabapentina tras la administración intravenosa de 150 mg es de 58±6 l (media ±DE). En pacientes con epilepsia, las concentraciones previas a la dosis en estado estacionario (Cmin) de gabapentina en el líquido cefalorraquídeo fueron aproximadamente el 20 % de las concentraciones plasmáticas correspondientes.
Eliminación
La gabapentina se elimina de la circulación sistémica por excreción renal como fármaco inalterado. La gabapentina no se metaboliza apreciablemente en humanos.
La vida media de eliminación de la gabapentina es de 5 a 7 horas y no se altera con la dosis o después de dosis múltiples. La tasa constante de eliminación de gabapentina, el aclaramiento plasmático y el aclaramiento renal son directamente proporcionales al aclaramiento de creatinina. En pacientes de edad avanzada y en pacientes con insuficiencia renal, el aclaramiento plasmático de gabapentina se reduce. La gabapentina se puede eliminar del plasma mediante hemodiálisis.
Poblaciones Específicas
Años
El efecto de la edad se estudió en sujetos de 20 a 80 años. El aclaramiento oral aparente (CL/F) de la gabapentina disminuyó a medida que aumentaba la edad, desde aproximadamente 225 ml/min en los menores de 30 años hasta aproximadamente 125 ml/min en los mayores de 70 años. El aclaramiento renal (CLr) y el CLr ajustado por área de superficie corporal también disminuyeron con la edad; sin embargo, la disminución del aclaramiento renal de gabapentina con la edad puede explicarse en gran medida por la disminución de la función renal. [ver DOSIFICACIÓN Y ADMINISTRACIÓN y Uso en poblaciones específicas ].
Género
Aunque no se ha realizado ningún estudio formal para comparar la farmacocinética de la gabapentina en hombres y mujeres, parece que los parámetros farmacocinéticos para hombres y mujeres son similares y no existen diferencias significativas de género.
La raza
No se han estudiado las diferencias farmacocinéticas debidas a la raza. Debido a que la gabapentina se excreta principalmente por vía renal y no existen diferencias raciales importantes en el aclaramiento de creatinina, no se esperan diferencias farmacocinéticas debidas a la raza.
Pediátrico
La farmacocinética de la gabapentina se determinó en 48 sujetos pediátricos con edades comprendidas entre 1 mes y 12 años después de una dosis de aproximadamente 10 mg/kg. Las concentraciones plasmáticas máximas fueron similares en todo el grupo de edad y ocurrieron de 2 a 3 horas después de la dosis. En general, los sujetos pediátricos entre 1 mes y
Se realizó un análisis farmacocinético poblacional en 253 sujetos pediátricos entre 1 mes y 13 años de edad. Los pacientes recibieron de 10 a 65 mg/kg/día administrados tres veces al día. El aclaramiento oral aparente (CL/F) fue directamente proporcional al aclaramiento de creatinina y esta relación fue similar después de una dosis única y en estado estacionario. Se observaron valores de depuración oral más altos en niños
Estos datos farmacocinéticos indican que la dosis diaria efectiva en pacientes pediátricos con epilepsia de 3 y 4 años de edad debe ser de 40 mg/kg/día para lograr concentraciones plasmáticas promedio similares a las alcanzadas en pacientes de 5 años y mayores que reciben 30 mg/día de gabapentina. kg/día [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
Pacientes adultos con insuficiencia renal
A los sujetos (N=60) con insuficiencia renal (depuración de creatinina media que oscilaba entre 13 y 114 ml/min) se les administraron dosis orales únicas de 400 mg de gabapentina. La semivida media de la gabapentina osciló entre aproximadamente 6,5 horas (pacientes con aclaramiento de creatinina >60 ml/min) y 52 horas (aclaramiento de creatinina 60 ml/min). grupo) a alrededor de 10 mL/min ( DOSIFICACIÓN Y ADMINISTRACIÓN y Uso en poblaciones específicas ]. No se han estudiado pacientes pediátricos con insuficiencia renal.
Hemodiálisis
En un estudio en sujetos adultos anúricos (N=11), la vida media de eliminación aparente de la gabapentina en días sin diálisis fue de aproximadamente 132 horas; durante la diálisis, la semivida aparente de la gabapentina se redujo a 3,8 horas. Por lo tanto, la hemodiálisis tiene un efecto significativo sobre la eliminación de gabapentina en sujetos anúricos [ver DOSIFICACIÓN Y ADMINISTRACIÓN y Uso en poblaciones específicas ].
Enfermedad Hepatica
Debido a que la gabapentina no se metaboliza, no se realizó ningún estudio en pacientes con insuficiencia hepática.
Interacciones con la drogas
Estudios in vitro
Se realizaron estudios in vitro para investigar el potencial de la gabapentina para inhibir las principales enzimas del citocromo P450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4) que median el metabolismo de fármacos y xenobióticos utilizando sustratos marcadores selectivos de isoformas y preparaciones microsomales de hígado humano. . Solo a la concentración más alta probada (171 mcg/mL; 1 mM) se observó un ligero grado de inhibición (14 % a 30 %) de la isoforma CYP2A6. No se observó inhibición de ninguna de las otras isoformas analizadas a concentraciones de gabapentina de hasta 171 mcg/mL (aproximadamente 15 veces la Cmax a 3600 mg/día) de la isoforma CYP2A6 observada. No se observó inhibición de ninguna de las otras isoformas probadas a concentraciones de gabapentina de hasta 171 mcg/mL (aproximadamente 15 veces la Cmax a 3600 mg/día).
Estudios in vivo
Los datos de interacciones medicamentosas descritos en esta sección se obtuvieron de estudios en adultos sanos y pacientes adultos con epilepsia.
fenitoína
En un estudio de dosis única (400 mg) y dosis múltiple (400 mg tres veces al día) de NEURONTIN 400 mg en pacientes epilépticos (N=8) mantenidos con monoterapia con fenitoína durante al menos 2 meses, la gabapentina no tuvo ningún efecto sobre el nivel mínimo en el estado estacionario. las concentraciones plasmáticas de fenitoína y fenitoína no tuvieron efecto sobre la farmacocinética de la gabapentina.
Carbamazepina
Las concentraciones plasmáticas mínimas de carbamazepina y carbamazepina 10, 11 en estado estacionario no se vieron afectadas por la administración concomitante de gabapentina (400 mg tres veces al día; N=12). Asimismo, la farmacocinética de la gabapentina no se vio alterada por la administración de carbamazepina.
Ácido valproico
Las concentraciones séricas mínimas medias de ácido valproico en estado estacionario antes y durante la administración concomitante de gabapentina (400 mg tres veces al día; N = 17) no fueron diferentes y los parámetros farmacocinéticos de la gabapentina tampoco se vieron afectados por el ácido valproico.
fenobarbital
Las estimaciones de los parámetros farmacocinéticos en estado estacionario para fenobarbital o gabapentina (300 mg tres veces al día; N = 12) son idénticas ya sea que los medicamentos se administren solos o juntos.
naproxeno
La coadministración (N=18) de cápsulas de naproxeno sódico (250 mg) con NEURONTIN (125 mg) parece aumentar la cantidad de gabapentina absorbida entre un 12 % y un 15 %. La gabapentina no tuvo efecto sobre los parámetros farmacocinéticos del naproxeno. Estas dosis son inferiores a las dosis terapéuticas de ambos fármacos. Se desconoce la magnitud de la interacción dentro de los rangos de dosis recomendados de cualquiera de los fármacos.
hidrocodona
La coadministración de NEURONTIN (125 a 500 mg; N=48) disminuye los valores de Cmax y AUC de hidrocodona (10 mg; N=50) de manera dependiente de la dosis en relación con la administración de hidrocodona sola; Los valores de Cmax y AUC son del 3 % al 4 % más bajos, respectivamente, después de la administración de 125 mg de NEURONTIN y del 21 % al 22 % más bajos, respectivamente, después de la administración de 500 mg de NEURONTIN. Se desconoce el mecanismo de esta interacción. La hidrocodona aumenta los valores del AUC de la gabapentina en un 14 %. Se desconoce la magnitud de la interacción a otras dosis.
Morfina
Un artículo de la literatura informó que cuando se administró una cápsula de morfina de liberación controlada de 60 mg 2 horas antes de una cápsula de NEURONTIN de 600 mg (N = 12), el AUC promedio de gabapentina aumentó en un 44 % en comparación con la administración de gabapentina sin morfina. Los valores de los parámetros farmacocinéticos de la morfina no se vieron afectados por la administración de NEURONTIN 2 horas después de la morfina. Se desconoce la magnitud de la interacción a otras dosis.
cimetidina
En presencia de cimetidina a 300 mg cuatro veces al día (N=12), el aclaramiento oral aparente medio de gabapentina disminuyó un 14 % y el aclaramiento de creatinina disminuyó un 10 %. Por lo tanto, la cimetidina pareció alterar la excreción renal tanto de gabapentina como de creatinina, un marcador endógeno de la función renal. No se espera que esta pequeña disminución en la excreción de gabapentina por cimetidina tenga importancia clínica. No se evaluó el efecto de la gabapentina sobre la cimetidina.
Anticonceptivo oral
Con base en el AUC y la vida media, los perfiles farmacocinéticos de dosis múltiples de noretindrona y etinilestradiol luego de la administración de tabletas que contenían 2.5 mg de acetato de noretindrona y 50 mcg de etinilestradiol fueron similares con y sin la administración conjunta de gabapentina (400 mg tres veces al día; N=13). La Cmax de noretindrona fue un 13 % mayor cuando se coadministró con gabapentina; no se espera que esta interacción tenga importancia clínica.
Antiácido (Maalox®) (hidróxido de aluminio, hidróxido de magnesio)
El antiácido (Maalox®) que contiene hidróxidos de magnesio y aluminio redujo la biodisponibilidad media de la gabapentina (N=16) en aproximadamente un 20 %. Esta disminución de la biodisponibilidad fue de alrededor del 10 % cuando se administró gabapentina 2 horas después de Maalox.
probenecid
El probenecid es un bloqueador de la secreción tubular renal. Los parámetros farmacocinéticos de gabapentina sin y con probenecid fueron comparables. Esto indica que la gabapentina no sufre secreción tubular renal por la vía que bloquea el probenecid.
Estudios clínicos
Neuralgia postherpética
NEURONTIN se evaluó para el tratamiento de la neuralgia posherpética (NPH) en dos estudios multicéntricos, aleatorizados, doble ciego, controlados con placebo. La población por intención de tratar (ITT) consistió en un total de 563 pacientes con dolor durante más de 3 meses después de la curación de la erupción cutánea por herpes zoster (Tabla 6).
TABLA 6. Estudios controlados de NPH: duración, dosis y número de pacientes
Cada estudio incluyó una fase doble ciego de 7 u 8 semanas (3 o 4 semanas de titulación y 4 semanas de dosis fija). Los pacientes iniciaron el tratamiento con titulación hasta un máximo de 900 mg/día de gabapentina durante 3 días. Luego, las dosis debían titularse en incrementos de 600 a 1200 mg/día a intervalos de 3 a 7 días hasta la dosis objetivo durante 3 a 4 semanas. Los pacientes registraron su dolor en un diario utilizando una escala numérica de calificación del dolor de 11 puntos que va de 0 (sin dolor) a 10 (el peor dolor posible). Se requirió una puntuación media de dolor durante la línea de base de al menos 4 para la aleatorización. Los análisis se realizaron utilizando la población ITT (todos los pacientes aleatorizados que recibieron al menos una dosis de la medicación del estudio).
Ambos estudios demostraron eficacia en comparación con el placebo en todas las dosis probadas.
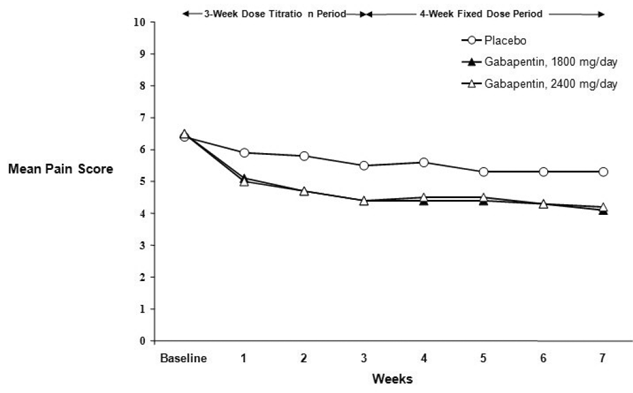
La reducción en las puntuaciones de dolor medias semanales se observó en la semana 1 en ambos estudios y se mantuvo hasta el final del tratamiento. Se observaron efectos de tratamiento comparables en todos los brazos de tratamiento activo. Los modelos farmacocinéticos/farmacodinámicos proporcionaron pruebas confirmatorias de la eficacia en todas las dosis. Las Figuras 1 y 2 muestran las puntuaciones de intensidad del dolor a lo largo del tiempo para los Estudios 1 y 2.
Figura 1. Puntuaciones medias semanales del dolor (casos observados en la población ITT): Estudio 1
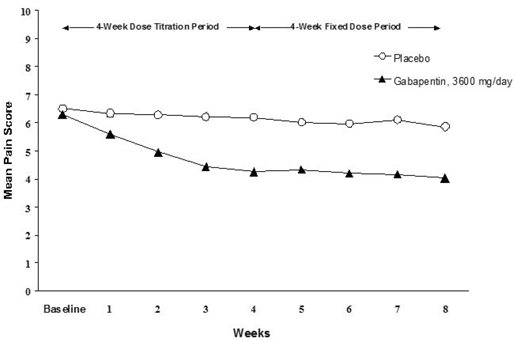
Figura 2. Puntuaciones medias semanales del dolor (casos observados en la población ITT): Estudio 2
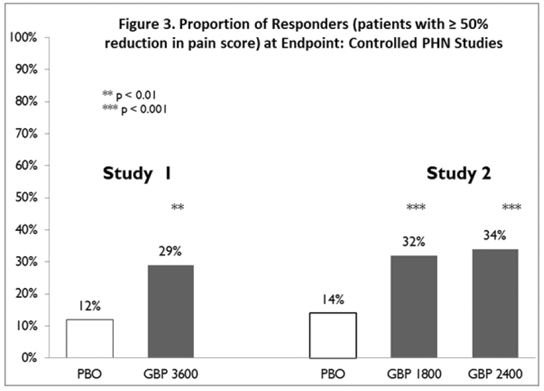
Se calculó la proporción de respondedores (aquellos pacientes que informaron una mejoría de al menos un 50 % en la puntuación final del dolor en comparación con el valor inicial) para cada estudio (Figura 3).
Figura 3. Proporción de respondedores (pacientes con ≥50 % de reducción en la puntuación del dolor) al final del estudio: estudios controlados de NPH
Epilepsia para convulsiones de inicio parcial (terapia complementaria)
La eficacia de NEURONTIN 400 mg como terapia adyuvante (añadido a otros fármacos antiepilépticos) se estableció en ensayos clínicos multicéntricos, controlados con placebo, doble ciego, de grupos paralelos en pacientes adultos y pediátricos (3 años y mayores) con convulsiones parciales refractarias.
Se obtuvo evidencia de eficacia en tres ensayos realizados en 705 pacientes (de 12 años de edad y mayores) y un ensayo realizado en 247 pacientes pediátricos (de 3 a 12 años de edad). Los pacientes inscritos tenían antecedentes de al menos 4 convulsiones parciales por mes a pesar de recibir uno o más medicamentos antiepilépticos a niveles terapéuticos y se observaron con su régimen establecido de medicamentos antiepilépticos durante un período inicial de 12 semanas (6 semanas en el estudio de pediatría). pacientes). En pacientes que continuaron teniendo al menos 2 (o 4 en algunos estudios) convulsiones por mes, se agregó NEURONTIN 300 mg o placebo a la terapia existente durante un período de tratamiento de 12 semanas. La eficacia se evaluó principalmente sobre la base del porcentaje de pacientes con una reducción del 50 % o más en la frecuencia de las convulsiones desde el inicio hasta el tratamiento (la "tasa de respuesta") y una medida derivada denominada tasa de respuesta, una medida de cambio definida como (T - B)/(T + B), donde B es la frecuencia de convulsiones de referencia del paciente y T es la frecuencia de convulsiones del paciente durante el tratamiento. La relación de respuesta se distribuye dentro del rango de -1 a +1. Un valor cero indica que no hay cambios, mientras que la eliminación completa de las convulsiones daría un valor de -1; el aumento de las tasas de convulsiones daría valores positivos. Una tasa de respuesta de -0,33 corresponde a una reducción del 50 % en la frecuencia de las crisis. Los resultados que se proporcionan a continuación son para todas las crisis parciales en la población por intención de tratar (todos los pacientes que recibieron cualquier dosis de tratamiento) en cada estudio, a menos que se indique lo contrario.
Un estudio comparó NEURONTIN 1200 mg/día, en tres dosis divididas con placebo. La tasa de respuesta fue del 23 % (14/61) en el grupo de NEURONTIN de 600 mg y del 9 % (6/66) en el grupo de placebo; la diferencia entre los grupos fue estadísticamente significativa. El índice de respuesta también fue mejor en el grupo NEURONTIN (-0,199) que en el grupo placebo (-0,044), una diferencia que también alcanzó significación estadística.
Un segundo estudio comparó principalmente NEURONTIN 1200 mg/día, en tres dosis divididas (N=101), con placebo (N=98). También se estudiaron grupos de dosis más pequeños de NEURONTIN de 300 mg (600 mg/día, N=53; 1800 mg/día, N=54) para obtener información sobre la respuesta a la dosis. La tasa de respuesta fue mayor en el grupo de NEURONTIN 1200 mg/día (16 %) que en el grupo de placebo (8 %), pero la diferencia no fue estadísticamente significativa. La tasa de respuesta a 600 mg (17 %) tampoco fue significativamente mayor que en el grupo de placebo, pero la tasa de respuesta en el grupo de 1800 mg (26 %) fue estadísticamente significativamente superior a la tasa de placebo. El índice de respuesta fue mejor en el grupo de NEURONTIN 1200 mg/día (-0,103) que en el grupo de placebo (-0,022); pero esta diferencia tampoco fue estadísticamente significativa (p = 0,224). Se observó una mejor respuesta en el grupo de NEURONTIN 600 mg/día (-0,105) y el grupo de 1800 mg/día (-0,222) que en el grupo de 1200 mg/día, alcanzando significación estadística el grupo de 1800 mg/día en comparación con el placebo grupo.
Un tercer estudio comparó NEURONTIN 900 mg/día, en tres dosis divididas (N=111) y placebo (N=109). Un grupo de dosificación adicional de NEURONTIN 1200 mg/día (N=52) proporcionó datos de respuesta a la dosis. Se observó una diferencia estadísticamente significativa en la tasa de respuesta en el grupo de NEURONTIN 900 mg/día (22 %) en comparación con el grupo de placebo (10 %). El índice de respuesta también fue significativamente superior desde el punto de vista estadístico en el grupo de NEURONTIN 900 mg/día (-0,119) en comparación con el grupo de placebo (-0,027), al igual que el índice de respuesta en NEURONTIN 1200 mg/día (-0,184) en comparación con el placebo.
También se realizaron análisis en cada estudio para examinar el efecto de NEURONTIN 300 mg en la prevención de convulsiones tónico-clónicas secundariamente generalizadas. Se incluyeron en estos análisis los pacientes que experimentaron una convulsión tónico-clónica secundariamente generalizada en la línea de base o en el período de tratamiento en los tres estudios controlados con placebo. Hubo varias comparaciones de índices de respuesta que mostraron una ventaja estadísticamente significativa para NEURONTIN en comparación con el placebo y tendencias favorables para casi todas las comparaciones.
El análisis de la tasa de respuesta utilizando datos combinados de los tres estudios y todas las dosis (N=162, NEURONTIN; N=89, placebo) también mostró una ventaja significativa de NEURONTIN sobre el placebo en la reducción de la frecuencia de crisis convulsivas tónico-clónicas secundariamente generalizadas.
En dos de los tres estudios controlados, se utilizó más de una dosis de NEURONTIN. Dentro de cada estudio, los resultados no mostraron un aumento constante de la respuesta a la dosis. Sin embargo, al observar los estudios, es evidente una tendencia hacia el aumento de la eficacia con el aumento de la dosis (consulte la Figura 4).
Figura 4. Tasa de respuesta en pacientes que recibieron NEURONTIN 600 mg expresada como diferencia del placebo por dosis y estudio: estudios de terapia adyuvante en pacientes ≥12 años de edad con convulsiones parciales
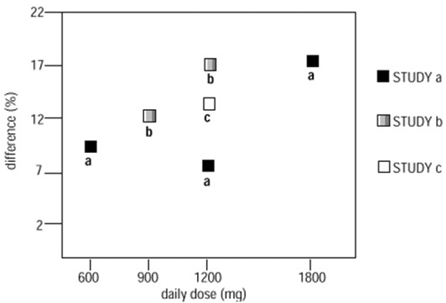
En la figura, la magnitud del efecto del tratamiento, medida en el eje Y en términos de la diferencia en la proporción de gabapentina y pacientes asignados a placebo que lograron una reducción del 50 % o más en la frecuencia de las convulsiones desde el inicio, se representa frente a la dosis diaria de gabapentina administrada. (eje X).
Aunque no se ha realizado un análisis formal por género, las estimaciones de respuesta (tasa de respuesta) derivadas de ensayos clínicos (398 hombres, 307 mujeres) indican que no existen diferencias importantes de género. No hubo un patrón consistente que indicara que la edad tuviera algún efecto sobre la respuesta a NEURONTIN. No hubo un número suficiente de pacientes de razas distintas a las caucásicas para permitir una comparación de la eficacia entre grupos raciales.
Un cuarto estudio en pacientes pediátricos de 3 a 12 años de edad comparó 25 a 35 mg/kg/día de NEURONTIN (N=118) con placebo (N=127). Para todas las convulsiones parciales en la población por intención de tratar, el índice de respuesta fue significativamente mejor desde el punto de vista estadístico para el grupo NEURONTIN (-0,146) que para el grupo placebo (-0,079). Para la misma población, la tasa de respuesta para NEURONTIN (21 %) no fue significativamente diferente de la del placebo (18 %).
Un estudio en pacientes pediátricos de 1 mes a 3 años de edad comparó 40 mg/kg/día de NEURONTIN (N=38) con placebo (N=38) en pacientes que estaban recibiendo al menos un fármaco antiepiléptico comercializado y tuvieron al menos una convulsión parcial durante el período de selección (dentro de las 2 semanas anteriores a la línea de base). Los pacientes tenían hasta 48 horas de línea de base y hasta 72 horas de monitoreo de EEG de video doble ciego para registrar y contar la aparición de convulsiones. No hubo diferencias estadísticamente significativas entre los tratamientos ni en la tasa de respuesta ni en la tasa de respondedores.
INFORMACIÓN DEL PACIENTE
NEURONTIN (Neu ron' tin) (Gabapentina) Cápsulas, tabletas y solución oral
¿Cuál es la información más importante que debo saber sobre NEURONTIN?
No deje de tomar NEURONTIN 300 mg sin antes hablar con su proveedor de atención médica.
La interrupción repentina de NEURONTIN puede causar problemas graves.
NEURONTIN 400 mg puede causar efectos secundarios graves que incluyen:
1. Pensamientos suicidas. Al igual que otros medicamentos antiepilépticos, NEURONTIN 100 mg puede causar pensamientos o acciones suicidas en un número muy pequeño de personas, aproximadamente 1 de cada 500.
Llame a un proveedor de atención médica de inmediato si tiene alguno de estos síntomas, especialmente si son nuevos, empeoran o le preocupan:
- pensamientos sobre el suicidio o la muerte
- intentos de suicidio
- depresión nueva o peor
- ansiedad nueva o peor
- sentirse agitado o inquieto
- ataques de pánico
- dificultad para dormir (insomnio)
- irritabilidad nueva o peor
- Actuar agresivo, estar enojado o violento.
- actuando sobre impulsos peligrosos
- un aumento extremo en la actividad y el habla (manía)
- otros cambios inusuales en el comportamiento o el estado de ánimo
¿Cómo puedo estar atento a los primeros síntomas de pensamientos y acciones suicidas?
- Preste atención a cualquier cambio, especialmente a los repentinos, en el estado de ánimo, el comportamiento, los pensamientos o los sentimientos.
- Mantenga todas las visitas de seguimiento con su proveedor de atención médica según lo programado.
Llame a su proveedor de atención médica entre visitas según sea necesario, especialmente si le preocupan los síntomas.
No deje de tomar NEURONTIN sin antes hablar con un proveedor de atención médica.
- La suspensión repentina de NEURONTIN 100 mg puede causar problemas graves. La interrupción repentina de un medicamento anticonvulsivo en un paciente que tiene epilepsia puede causar convulsiones que no se detendrán (estado epiléptico).
- Los pensamientos o acciones suicidas pueden ser causados por otras cosas además de los medicamentos. Si tiene pensamientos o acciones suicidas, su proveedor de atención médica puede buscar otras causas.
2. Cambios en el comportamiento y el pensamiento - El uso de NEURONTIN 100 mg en niños de 3 a 12 años puede causar cambios emocionales, comportamiento agresivo, problemas de concentración, inquietud, cambios en el rendimiento escolar e hiperactividad.
3. NEURONTIN puede causar reacciones alérgicas graves o potencialmente mortales que pueden afectar su piel u otras partes de su cuerpo, como el hígado o las células sanguíneas. Esto puede ocasionar que sea hospitalizado o que deje de tomar NEURONTIN. Puede o no tener una erupción con una reacción alérgica causada por NEURONTIN. Llame a un proveedor de atención médica de inmediato si tiene alguno de los siguientes síntomas:
- erupción cutanea
- urticaria
- respiración dificultosa
- fiebre
- glándulas inflamadas que no desaparecen
- hinchazón de la cara, los labios, la garganta o la lengua
- coloración amarillenta de la piel o del blanco de los ojos
- hematomas o sangrado inusuales
- fatiga severa o debilidad
- dolor muscular inesperado
- infecciones frecuentes
Estos síntomas pueden ser los primeros signos de una reacción grave. Un proveedor de atención médica debe examinarlo para decidir si debe continuar tomando NEURONTIN.
4. Problemas respiratorios graves. Se pueden producir problemas respiratorios graves cuando NEURONTIN se toma con otros medicamentos que pueden causar somnolencia intensa o disminución de la conciencia, o cuando lo toma alguien que ya tiene problemas respiratorios. Tenga cuidado con el aumento de la somnolencia o la disminución de la respiración cuando comience NEURONTIN 300 mg o cuando aumente la dosis. Obtenga ayuda de inmediato si se presentan problemas respiratorios.
¿Qué es NEURONTIN 400mg?
NEURONTIN 400 mg es un medicamento recetado que se usa para tratar:
- Dolor de los nervios dañados (dolor posherpético) que sigue a la cicatrización de la culebrilla (una erupción dolorosa que se presenta después de una infección por herpes zoster) en adultos.
- Convulsiones parciales cuando se toman junto con otros medicamentos en adultos y niños mayores de 3 años con convulsiones.
¿Quién no debe tomar NEURONTIN?
No tome NEURONTIN si es alérgico a la gabapentina oa cualquiera de los demás componentes de NEURONTIN. Consulte el final de esta Guía del medicamento para obtener una lista completa de los ingredientes de NEURONTIN.
¿Qué debo decirle a mi proveedor de atención médica antes de tomar NEURONTIN 300 mg?
Antes de tomar NEURONTIN 400 mg, informe a su proveedor de atención médica si:
- tiene o ha tenido problemas renales o está en hemodiálisis
- tiene o ha tenido depresión, problemas del estado de ánimo o pensamientos o conductas suicidas
- tener diabetes
- tiene problemas respiratorios
- está embarazada o planea quedar embarazada. No se sabe si NEURONTIN puede dañar a su bebé nonato. Informe a su proveedor de atención médica de inmediato si queda embarazada mientras toma NEURONTIN. Usted y su proveedor de atención médica decidirán si debe tomar NEURONTIN 100 mg durante el embarazo. Registro de Embarazo: Si queda embarazada mientras toma NEURONTIN, hable con su proveedor de atención médica acerca de registrarse en el Registro de Embarazo de Medicamentos Antiepilépticos de América del Norte (NAAED). El propósito de este registro es recopilar información sobre la seguridad de los medicamentos antiepilépticos durante el embarazo. Puede inscribirse en este registro llamando al 1-888-233-2334.
- está amamantando o planea amamantar. NEURONTIN puede pasar a la leche materna. Usted y su proveedor de atención médica deben decidir cómo alimentará a su bebé mientras toma NEURONTIN.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluyendo medicamentos recetados y de venta libre, vitaminas y suplementos a base de hierbas. Informe especialmente a su proveedor de atención médica si toma algún analgésico opioide (como la oxicodona), cualquier medicamento para la ansiedad (como el lorazepam) o el insomnio (como el zolpidem), o cualquier medicamento que le dé sueño.
Puede tener una mayor probabilidad de mareos, somnolencia o problemas respiratorios si estos medicamentos se toman con NEURONTIN.
Tomar NEURONTIN 100 mg con otros medicamentos puede causar efectos secundarios o afectar su funcionamiento. No empiece ni suspenda otros medicamentos sin hablar con su proveedor de atención médica.
Conozca los medicamentos que toma. Mantenga una lista de ellos y muéstresela a su proveedor de atención médica y farmacéutico cuando obtenga un nuevo medicamento.
¿Cómo debo tomar NEURONTIN 400mg?
- Tome NEURONTIN exactamente según lo prescrito. Su proveedor de atención médica le dirá cuánto NEURONTIN 600 mg debe tomar.
- No cambie su dosis de NEURONTIN sin hablar con su proveedor de atención médica.
- Si toma comprimidos de NEURONTIN y parte un comprimido por la mitad, la mitad restante del comprimido debe tomarse en su próxima dosis programada. La mitad de las tabletas que no se usen dentro de los 28 días posteriores a la ruptura deben desecharse.
- Tome las cápsulas de NEURONTIN con agua.
- Los comprimidos de NEURONTIN se pueden tomar con o sin alimentos. Si toma un antiácido que contiene aluminio y magnesio, como Maalox®, Mylanta®, Gelusil®, Gaviscon® o Di-Gel®, debe esperar al menos 2 horas antes de tomar su próxima dosis de NEURONTIN. Si toma demasiado NEURONTIN 600 mg, llame a su proveedor de atención médica o al Centro de control de intoxicaciones local de inmediato al 1-800-222-1222.
¿Qué debo evitar mientras tomo NEURONTIN?
- No beba alcohol ni tome otros medicamentos que le causen sueño o mareos mientras toma NEURONTIN sin antes hablar con su proveedor de atención médica. Tomar NEURONTIN 600 mg con alcohol o drogas que causen somnolencia o mareos puede empeorar su somnolencia o mareos.
- No conduzca, opere maquinaria pesada ni realice otras actividades peligrosas hasta que sepa cómo le afecta NEURONTIN 100 mg. NEURONTIN 600 mg puede ralentizar su pensamiento y sus habilidades motoras.
¿Cuáles son los posibles efectos secundarios de NEURONTIN 300mg?
NEURONTIN 100 mg puede causar efectos secundarios graves que incluyen:
Ver “¿Cuál es la información más importante que debo saber sobre NEURONTIN?”
- problemas para conducir mientras usa NEURONTIN. Consulte "¿Qué debo evitar mientras tomo Neurontin 600 mg?"
- somnolencia y mareos, que podrían aumentar la ocurrencia de lesiones accidentales, incluidas caídas
- Los efectos secundarios más comunes de NEURONTIN 100 mg incluyen:
- falta de cordinacion
- infección viral
- sentirse somnoliento
- náuseas y vómitos
- dificultad para hablar
- temblor
- hinchazón, generalmente de piernas y pies
- sensación de cansancio
- fiebre
- movimientos bruscos
- dificultad con la coordinación
- visión doble
- movimiento inusual de los ojos
Informe a su proveedor de atención médica si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de NEURONTIN. Para obtener más información, consulte a su proveedor de atención médica o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
¿Cómo debo almacenar NEURONTIN?
- Guarde NEURONTIN 400 mg en cápsulas y tabletas entre 20 °C y 25 °C (68 °F y 77 °F).
- Guarde NEURONTIN 400 mg Solución oral en el refrigerador entre 36 °F y 46 °F (2 °C y 8 °C).
Mantenga NEURONTIN 600 mg y todos los medicamentos fuera del alcance de los niños.
Información general sobre el uso seguro y eficaz de NEURONTIN
veces, los medicamentos se recetan para fines distintos a los enumerados en la Guía del medicamento. No use NEURONTIN 300 mg para una condición para la cual no fue recetado. No administre NEURONTIN a otras personas, incluso si tienen los mismos síntomas que usted. Puede que les haga daño.
Esta Guía del medicamento resume la información más importante sobre NEURONTIN. Si desea obtener más información, hable con su proveedor de atención médica. Puede pedirle a su proveedor de atención médica o farmacéutico información sobre NEURONTIN 100 mg que se escribió para profesionales de la salud.
Para obtener más información, visite http://www.pfizer.com o llame al 1-800-438-1985.
¿Cuáles son los ingredientes de NEURONTIN 400 mg?
Ingrediente activo: gabapentina
Ingredientes inactivos en las cápsulas: lactosa, almidón de maíz, talco, gelatina, dióxido de titanio y FD&C Blue No. 2.
La cubierta de la cápsula de 300 mg también contiene: óxido de hierro amarillo.
La cubierta de la cápsula de 400 mg también contiene: óxido de hierro rojo y óxido de hierro amarillo.
Ingredientes inactivos en las tabletas: poloxámero 407, copovidona, maicena, estearato de magnesio, hidroxipropilcelulosa, talco y cera de candelilla
Ingredientes inactivos en la solución oral: glicerina, xilitol, agua purificada y sabor artificial.
Esta Guía del medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de EE. UU.