Voltaren 100mg, 50mg Diclofenac Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Voltaren y cómo se usa?
Voltaren se usa para tratar los síntomas de dolor asociados con la artritis reumatoide, la osteoartritis, la espondilitis anquilosante, la dismenorrea y el dolor de leve a moderado. Voltaren 100 mg se puede usar solo o con otros medicamentos.
Voltaren pertenece a una clase de medicamentos llamados medicamentos antiinflamatorios no esteroideos (AINE).
¿Cuáles son los posibles efectos secundarios de Voltaren 50 mg?
Voltaren puede causar efectos secundarios graves que incluyen:
- dolor de cabeza,
- hambre,
- transpiración,
- irritabilidad,
- mareo,
- náuseas,
- frecuencia cardíaca rápida,
- sentirse ansioso o tembloroso,
- entumecimiento u hormigueo en sus manos, brazos, piernas o pies,
- debilidad en sus brazos, manos, piernas o pies,
- dolor ardiente en sus brazos, manos, piernas o pies,
- cambios graves de humor o de comportamiento,
- nerviosismo,
- confusión,
- agitación,
- paranoia,
- alucinaciones,
- problemas de memoria,
- problemas para concentrarse,
- pensamientos de suicidio,
- ruptura de tendón,
- dolor súbito,
- hinchazón,
- hematomas,
- sensibilidad,
- rigidez,
- problemas de movimiento,
- un sonido de chasquido o estallido en cualquiera de sus articulaciones,
- dolor de estómago severo,
- diarrea acuosa o con sangre,
- revoloteando en tu pecho,
- dificultad para respirar,
- erupción cutanea,
- problemas respiratorios,
- ataques (convulsiones),
- dolores de cabeza severos,
- problemas de la vista,
- dolor detrás de tus ojos,
- dolor de estómago superior,
- pérdida de apetito,
- orina oscura,
- heces de color arcilla, y
- coloración amarillenta de la piel o los ojos (ictericia)
Pida ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Voltaren incluyen:
- indigestión,
- gas,
- dolor de estómago,
- náuseas,
- vómitos,
- Diarrea,
- estreñimiento,
- dolor de cabeza,
- mareo,
- somnolencia,
- congestión nasal,
- Comezón,
- aumento de la sudoración,
- aumento de la presión arterial, y
- hinchazón o dolor en sus brazos o piernas
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Voltaren. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
ADVERTENCIA
HEPATOTOXICIDAD, TOXICIDAD CARDÍACA, TOXICIDAD EMBRIOFETAL
Eventos trombóticos cardiovasculares
- Los medicamentos antiinflamatorios no esteroideos (AINE) aumentan el riesgo de eventos trombóticos cardiovasculares graves, incluidos infarto de miocardio y accidente cerebrovascular, que pueden ser fatales. Este riesgo puede ocurrir temprano en el tratamiento y puede aumentar con la duración del uso (ver ADVERTENCIAS).
- VOLTAREN® está contraindicado en el marco de una cirugía de injerto de bypass de arteria coronaria (CABG) (ver CONTRAINDICACIONES, ADVERTENCIAS).
Sangrado gastrointestinal, ulceración y perforación
- Los AINE causan un mayor riesgo de eventos adversos gastrointestinales (GI) graves que incluyen sangrado, ulceración y perforación del estómago o los intestinos, que pueden ser fatales. Estos eventos pueden ocurrir en cualquier momento durante el uso y sin síntomas de advertencia. Los pacientes de edad avanzada y los pacientes con antecedentes de enfermedad de úlcera péptica y/o hemorragia gastrointestinal tienen un mayor riesgo de eventos gastrointestinales graves. (ver ADVERTENCIAS).
DESCRIPCIÓN
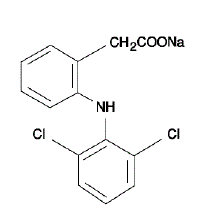
VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) es un derivado del ácido bencenoacético. VOLTAREN 50 mg está disponible en comprimidos de liberación retardada (recubrimiento entérico) de 75 mg (rosa claro) para administración oral. El diclofenaco sódico es un polvo cristalino blanco o ligeramente amarillento y es poco soluble en agua a 25°C. El nombre químico es sal monosódica del ácido 2-[(2,6-diclorofenil)amino]bencenoacético. El peso molecular es 318,14. Su fórmula molecular es C14H10Cl2NNaO2, y tiene la siguiente fórmula estructural
Los ingredientes inactivos de VOLTAREN incluyen: hidroxipropilmetilcelulosa, óxido de hierro, lactosa, estearato de magnesio, copolímero de ácido metacrílico, celulosa microcristalina, polietilenglicol, povidona, propilenglicol, hidróxido de sodio, glicolato de almidón de sodio, talco, dióxido de titanio.
INDICACIONES
Considere detenidamente los posibles beneficios y riesgos de VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) y otras opciones de tratamiento antes de decidir usar VOLTAREN. Utilice la dosis eficaz más baja durante el menor tiempo posible de acuerdo con los objetivos de tratamiento de cada paciente (ver ADVERTENCIAS ; Hemorragia gastrointestinal , Ulceración , y Perforación ).
VOLTAREN está indicado:
- para el alivio de los signos y síntomas de la osteoartritis
- para el alivio de los signos y síntomas de la artritis reumatoide
- para uso agudo o a largo plazo en el alivio de los signos y síntomas de la espondilitis anquilosante
DOSIFICACIÓN Y ADMINISTRACIÓN
Considere detenidamente los posibles beneficios y riesgos de VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) y otras opciones de tratamiento antes de decidir usar VOLTAREN. Utilice la dosis eficaz más baja durante el menor tiempo posible de acuerdo con los objetivos de tratamiento de cada paciente (ver ADVERTENCIAS ; Sangrado gastrointestinal, ulceración y perforación ).
Después de observar la respuesta a la terapia inicial con VOLTAREN 100 mg, la dosis y la frecuencia deben ajustarse para adaptarse a las necesidades individuales de cada paciente.
Para el alivio de la osteoartritis, la dosis recomendada es de 100 a 150 mg/día en dosis divididas (50 mg dos veces al día o tres veces al día, o 75 mg dos veces al día).
Para el alivio de la artritis reumatoide, la dosis recomendada es de 150 a 200 mg/día en dosis divididas (50 mg tres veces al día o cuatro veces al día, o 75 mg dos veces al día).
Para el alivio de la espondilitis anquilosante, la dosis recomendada es de 100 a 125 mg/día, administrados en dosis de 25 mg cuatro veces al día, con una dosis adicional de 25 mg al acostarse si es necesario.
Diferentes formulaciones de diclofenaco [VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico); VOLTAREN®ÂXR (tabletas de liberación prolongada de diclofenaco sódico); CATAFLAM® (tabletas de liberación inmediata de diclofenaco potásico)] no son necesariamente bioequivalentes incluso si la concentración de miligramos es la misma.
CÓMO SUMINISTRADO
VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) - 75 miligramos
Comprimidos de color rosa claro, biconvexos, de forma triangular, con cubierta entérica (con la inscripción “VOLTAREN 75” en una cara en tinta negra)
Botellas de 100 CDN 0028-0264-01
Almacenar a temperatura ambiente de 20 °C a 25 °C (68 °F a 77 °F); excursiones permitidas entre 15°C y 30°C (59°F y 86°F) [ver Temperatura ambiente controlada por USP ].
Proteger de la humedad. Dispensar en recipiente hermético (USP).
Distribuido por: Novartis Pharmaceuticals Corporation East Hanover, NJ 07936. Revisado: abril de 2021
EFECTOS SECUNDARIOS
Las siguientes reacciones adversas se analizan con mayor detalle en otras secciones de la etiqueta:
- Eventos trombóticos cardiovasculares (ver ADVERTENCIAS )
- Sangrado gastrointestinal, ulceración y perforación (ver ADVERTENCIAS )
- Hepatotoxicidad (ver ADVERTENCIAS )
- Hipertensión (ver ADVERTENCIAS )
- Insuficiencia cardíaca y edema (ver ADVERTENCIAS )
- Toxicidad renal e hiperpotasemia (ver ADVERTENCIAS )
- Reacciones anafilácticas (ver ADVERTENCIAS )
- Reacciones cutáneas graves (ver ADVERTENCIAS )
- Toxicidad hematológica (ver ADVERTENCIAS )
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica.
En pacientes que toman VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) u otros AINE, las experiencias adversas informadas con mayor frecuencia que ocurren en aproximadamente el 1% al 10% de los pacientes son: Experiencias gastrointestinales, que incluyen: dolor abdominal, estreñimiento, diarrea, dispepsia, flatulencia, hemorragia grave/perforación, acidez estomacal, náuseas, úlceras gastrointestinales (gástricas/duodenales) y vómitos.
Función renal anormal, anemia, mareos, edema, enzimas hepáticas elevadas, dolores de cabeza, aumento del tiempo de sangrado, prurito, erupciones cutáneas y tinnitus. Las experiencias adversas adicionales informadas ocasionalmente incluyen:
Cuerpo como un todo: fiebre, infección, sepsis
Sistema cardiovascular: insuficiencia cardíaca congestiva, hipertensión, taquicardia, síncope
Sistema digestivo: boca seca, esofagitis, úlceras gástricas/pépticas, gastritis, sangrado gastrointestinal, glositis, hematemesis, hepatitis, ictericia
Sistema Hemático y Linfático: equimosis, eosinofilia, leucopenia, melena, púrpura, rectorragia, estomatitis, trombocitopenia
Metabólicas y Nutricionales: cambios de peso
Sistema nervioso: ansiedad, astenia, confusión, depresión, alteraciones del sueño, somnolencia, insomnio, malestar general, nerviosismo, parestesia, somnolencia, temblores, vértigo
Sistema respiratorio: asma, disnea
Piel y apéndices: alopecia, fotosensibilidad, aumento de la sudoración
Sentidos especiales: visión borrosa
Sistema urogenital: cistitis, disuria, hematuria, nefritis intersticial, oliguria/poliuria, proteinuria, insuficiencia renal
Otras reacciones adversas, que ocurren raramente son:
Cuerpo como un todo: reacciones anafilácticas, cambios en el apetito, muerte
Sistema cardiovascular: arritmia, hipotensión, infarto de miocardio, palpitaciones, vasculitis
Sistema digestivo: colitis, eructos, hepatitis fulminante con y sin ictericia, insuficiencia hepática, necrosis hepática, pancreatitis
Sistema Hemático y Linfático: agranulocitosis, anemia hemolítica, anemia aplásica, linfadenopatía, pancitopenia
Metabólicas y Nutricionales: hiperglucemia
Sistema nervioso: convulsiones, coma, alucinaciones, meningitis
Sistema respiratorio: depresión respiratoria, neumonía
Piel y apéndices: angioedema, necrólisis epidérmica tóxica, eritema multiforme, dermatitis exfoliativa, síndrome de Stevens-Johnson, urticaria
Sentidos especiales: conjuntivitis, deficiencia auditiva
SOBREDOSIS
Los síntomas que siguen a las sobredosis agudas de AINE generalmente se han limitado a letargo, somnolencia, náuseas, vómitos y dolor epigástrico, que generalmente han sido reversibles con atención de apoyo. Ha ocurrido sangrado gastrointestinal. Se han producido hipertensión, insuficiencia renal aguda, depresión respiratoria y coma, pero fueron raros (ver ADVERTENCIAS ; Eventos trombóticos cardiovasculares, hemorragia gastrointestinal, ulceración e hipertensión, toxicidad renal e hiperpotasemia ).
Maneje a los pacientes con atención sintomática y de apoyo después de una sobredosis de AINE. No hay antídotos específicos. Considere emesis y/o carbón activado (60 a 100 gramos en adultos, 1 a 2 gramos por kg de peso corporal en pacientes pediátricos) y/o catártico osmótico en pacientes sintomáticos vistos dentro de las cuatro horas posteriores a la ingestión o en pacientes con una sobredosis grande ( 5 a 10 veces la dosis recomendada). La diuresis forzada, la alcalinización de la orina, la hemodiálisis o la hemoperfusión pueden no ser útiles debido a la alta unión a proteínas.
Para obtener información adicional sobre el tratamiento de sobredosis, comuníquese con un centro de control de intoxicaciones (1-800-222-1222).
INTERACCIONES CON LA DROGAS
Consulte la Tabla 2 para conocer las interacciones farmacológicas clínicamente significativas con diclofenaco.
ADVERTENCIAS
Eventos trombóticos cardiovasculares
Los ensayos clínicos de varios AINE selectivos y no selectivos de COX-2 de hasta 3 años de duración han mostrado un mayor riesgo de eventos trombóticos cardiovasculares (CV) graves, incluido infarto de miocardio (IM) y accidente cerebrovascular, que pueden ser fatales. Según los datos disponibles, no está claro que el riesgo de eventos trombóticos cardiovasculares sea similar para todos los AINE. El aumento relativo de eventos trombóticos cardiovasculares graves con respecto al inicio conferido por el uso de AINE parece ser similar en aquellos con y sin enfermedad cardiovascular conocida o factores de riesgo de enfermedad cardiovascular. Sin embargo, los pacientes con enfermedad cardiovascular conocida o factores de riesgo tuvieron una incidencia absoluta más alta de eventos trombóticos cardiovasculares graves en exceso, debido a su mayor tasa inicial. Algunos estudios observacionales encontraron que este aumento del riesgo de eventos trombóticos cardiovasculares graves comenzó ya en las primeras semanas de tratamiento. El aumento del riesgo trombótico CV se ha observado de forma más consistente con dosis más altas.
Para minimizar el riesgo potencial de un evento cardiovascular adverso en pacientes tratados con AINE, use la dosis efectiva más baja durante el menor tiempo posible. Los médicos y los pacientes deben permanecer atentos al desarrollo de tales eventos durante todo el curso del tratamiento, incluso en ausencia de síntomas CV previos. Se debe informar a los pacientes sobre los síntomas de eventos CV graves y los pasos a seguir si ocurren.
No existe evidencia consistente de que el uso concurrente de aspirina mitigue el mayor riesgo de eventos trombóticos cardiovasculares graves asociados con el uso de AINE. El uso simultáneo de aspirina y un AINE, como el diclofenaco, aumenta el riesgo de eventos gastrointestinales (GI) graves (ver ADVERTENCIAS ; Hemorragia gastrointestinal , Ulceración , y Perforación ).
Estado posterior a la cirugía de injerto de derivación de la arteria coronaria (CABG)
Dos grandes ensayos clínicos controlados de un AINE selectivo de COX-2 para el tratamiento del dolor en los primeros 10 a 14 días posteriores a la cirugía CABG encontraron una mayor incidencia de infarto de miocardio y accidente cerebrovascular. Los AINE están contraindicados en el contexto de CABG (ver CONTRAINDICACIONES ).
Pacientes post-IM
Los estudios observacionales realizados en el Registro Nacional Danés han demostrado que los pacientes tratados con AINE en el período posterior al IM tenían un mayor riesgo de reinfarto, muerte relacionada con CV y mortalidad por todas las causas a partir de la primera semana de tratamiento. En esta misma cohorte, la incidencia de muerte en el primer año posterior al IM fue de 20 por 100 años-persona en pacientes tratados con AINE en comparación con 12 por 100 años-persona en pacientes no expuestos a AINE. Aunque la tasa absoluta de muerte disminuyó un poco después del primer año posterior al IM, el aumento del riesgo relativo de muerte en los usuarios de AINE persistió durante al menos los siguientes cuatro años de seguimiento.
Evite el uso de VOLTAREN en pacientes con un infarto de miocardio reciente a menos que se espere que los beneficios superen el riesgo de eventos trombóticos cardiovasculares recurrentes. Si se usa VOLTAREN en pacientes con un infarto de miocardio reciente, controle a los pacientes para detectar signos de isquemia cardíaca.
Sangrado gastrointestinal, ulceración y perforación
Los AINE, incluido el diclofenaco, causan eventos adversos gastrointestinales (GI) graves, que incluyen inflamación, sangrado, ulceración y perforación del esófago, el estómago, el intestino delgado o el intestino grueso, que pueden ser fatales. Estos eventos adversos graves pueden ocurrir en cualquier momento, con o sin síntomas de advertencia, en pacientes tratados con AINE. Solo uno de cada cinco pacientes, que desarrollan un evento adverso GI superior grave en la terapia con AINE, es sintomático. Se produjeron úlceras gastrointestinales superiores, sangrado macroscópico o perforación causados por AINE en aproximadamente el 1 % de los pacientes tratados durante 3 a 6 meses, y en aproximadamente el 2 % al 4 % de los pacientes tratados durante un año. Sin embargo, incluso la terapia a corto plazo no está exenta de riesgos.
Factores de riesgo de sangrado, ulceración y perforación GI
Los pacientes con antecedentes de enfermedad de úlcera péptica y/o sangrado GI que usan AINE tenían un riesgo más de 10 veces mayor de desarrollar sangrado GI en comparación con los pacientes sin estos factores de riesgo. Otros factores que aumentan el riesgo de hemorragia GI en pacientes tratados con NSAID incluyen una duración más prolongada de la terapia con NSAID, el uso concomitante de corticosteroides orales, aspirina, anticoagulantes o inhibidores selectivos de la recaptación de serotonina (ISRS); tabaquismo, consumo de alcohol, edad avanzada y mal estado general de salud. La mayoría de los informes posteriores a la comercialización de eventos GI fatales ocurrieron en pacientes ancianos o debilitados. Además, los pacientes con enfermedad hepática avanzada y/o coagulopatía tienen un mayor riesgo de hemorragia gastrointestinal.
Estrategias PARA MINIMIZAR LOS RIESGOS GI EN PACIENTES TRATADOS CON AINE
- Use la dosis efectiva más baja durante el menor tiempo posible.
- Evite la administración de más de un AINE a la vez
- Evite el uso en pacientes con mayor riesgo a menos que se espere que los beneficios superen el mayor riesgo de hemorragia. Para tales pacientes, así como para aquellos con sangrado GI activo, considere terapias alternativas distintas a los AINE.
- Permanezca alerta a los signos y síntomas de ulceración gastrointestinal y sangrado durante la terapia con AINE.
- Si se sospecha un evento adverso GI grave, inicie de inmediato la evaluación y el tratamiento, y suspenda VOLTAREN 50 mg hasta que se descarte un evento adverso GI grave.
- En el contexto del uso concomitante de aspirina en dosis bajas para la profilaxis cardíaca, controle más de cerca a los pacientes para detectar evidencia de hemorragia GI (ver PRECAUCIONES ; Interacciones con la drogas ).
hepatotoxicidad
En ensayos clínicos de productos que contienen diclofenaco, se observaron elevaciones significativas (es decir, más de 3 veces el límite superior normal [ULN]) de aspartato aminotransferasa (AST) (también conocida como SGOT) en aproximadamente el 2% de aproximadamente 5700 pacientes en algún tiempo durante el tratamiento con diclofenaco (no se midió la alanina aminotransferasa [ALT] en todos los estudios).
En un gran ensayo controlado abierto de 3700 pacientes tratados con diclofenaco sódico oral durante 2 a 6 meses, los pacientes fueron monitoreados primero a las 8 semanas y 1200 pacientes fueron monitoreados nuevamente a las 24 semanas. Se produjeron elevaciones significativas de ALT y/o AST en aproximadamente el 4% de los pacientes e incluyeron elevaciones marcadas (más de 8 veces el ULN) en aproximadamente el 1% de los 3700 pacientes. En ese estudio abierto, se observó una mayor incidencia de elevaciones limítrofes (menos de 3 veces el ULN), moderadas (3 a 8 veces el ULN) y marcadas (más de 8 veces el ULN) de ALT o AST en pacientes recibir diclofenaco en comparación con otros AINE. Las elevaciones de las transaminasas se observaron con mayor frecuencia en pacientes con osteoartritis que en aquellos con artritis reumatoide.
Casi todas las elevaciones significativas de las transaminasas se detectaron antes de que los pacientes presentaran síntomas. Ocurrieron pruebas anormales durante los primeros 2 meses de terapia con diclofenaco en 42 de los 51 pacientes en todos los ensayos que desarrollaron elevaciones marcadas de transaminasas.
En informes posteriores a la comercialización, se informaron casos de hepatotoxicidad inducida por fármacos en el primer mes y, en algunos casos, en los primeros 2 meses de terapia, pero puede ocurrir en cualquier momento durante el tratamiento con diclofenaco. La vigilancia posterior a la comercialización ha informado casos de reacciones hepáticas graves, que incluyen necrosis hepática, ictericia, hepatitis fulminante con y sin ictericia e insuficiencia hepática. Algunos de estos casos informados resultaron en muertes o trasplantes de hígado.
En un estudio europeo retrospectivo de casos y controles, basado en la población, 10 casos de lesión hepática inducida por fármacos asociada con diclofenaco con el uso actual en comparación con la ausencia de uso de diclofenaco se asociaron con una razón de probabilidad estadísticamente significativa ajustada al cuádruple de lesión hepática. En este estudio en particular, basado en un número total de 10 casos de daño hepático asociado con diclofenaco, la razón de probabilidad ajustada aumentó aún más con el sexo femenino, las dosis de 150 mg o más y la duración del uso durante más de 90 días.
Los médicos deben medir las transaminasas al inicio y periódicamente en pacientes que reciben terapia a largo plazo con diclofenaco, porque se puede desarrollar una hepatotoxicidad grave sin un pródromo de síntomas distintivos. No se conocen los tiempos óptimos para realizar la primera y posteriores mediciones de transaminasas. Según los datos de ensayos clínicos y las experiencias posteriores a la comercialización, las transaminasas deben controlarse dentro de las 4 a 8 semanas posteriores al inicio del tratamiento con diclofenaco. Sin embargo, pueden ocurrir reacciones hepáticas graves en cualquier momento durante el tratamiento con diclofenaco.
Si las pruebas hepáticas anormales persisten o empeoran, si se desarrollan signos y/o síntomas clínicos consistentes con una enfermedad hepática, o si ocurren manifestaciones sistémicas (p. ej., eosinofilia, erupción cutánea, dolor abdominal, diarrea, orina oscura, etc.), VOLTAREN debe suspenderse de inmediato. .
Informar a los pacientes sobre los signos y síntomas de advertencia de hepatotoxicidad (p. ej., náuseas, fatiga, letargo, diarrea, prurito, ictericia, sensibilidad en el cuadrante superior derecho y síntomas “similares a la gripe”). Si se desarrollan signos y síntomas clínicos consistentes con una enfermedad hepática, o si ocurren manifestaciones sistémicas (p. ej., eosinofilia, sarpullido, etc.), suspenda VOLTAREN de inmediato y realice una evaluación clínica del paciente.
Para minimizar el riesgo potencial de un evento hepático adverso en pacientes tratados con VOLTAREN, use la dosis efectiva más baja durante el menor tiempo posible. Tenga cuidado al prescribir VOLTAREN 100 mg con medicamentos concomitantes que se sabe que son potencialmente hepatotóxicos (p. ej., paracetamol, antibióticos, antiepilépticos).
Hipertensión
Los AINE, incluido VOLTAREN 100 mg, pueden provocar una nueva aparición de hipertensión o un empeoramiento de la hipertensión preexistente, cualquiera de los cuales puede contribuir al aumento de la incidencia de eventos cardiovasculares. Los pacientes que toman inhibidores de la enzima convertidora de angiotensina (ECA), diuréticos tiazídicos o diuréticos de asa pueden tener una respuesta deficiente a estas terapias cuando toman AINE (ver PRECAUCIONES ; INTERACCIONES CON LA DROGAS ).
Controle la presión arterial (PA) durante el inicio del tratamiento con AINE y durante el curso de la terapia.
Insuficiencia cardíaca y edema
El metanálisis de ensayos controlados aleatorios de Coxib y NSAID Trialists' Collaboration demostró un aumento de aproximadamente 2 veces en la hospitalización por insuficiencia cardíaca en pacientes tratados selectivamente con COX-2 y pacientes tratados con AINE no selectivos en comparación con pacientes tratados con placebo. En un estudio del Registro Nacional Danés de pacientes con insuficiencia cardíaca, el uso de AINE aumentó el riesgo de infarto de miocardio, hospitalización por insuficiencia cardíaca y muerte.
Además, se ha observado retención de líquidos y edema en algunos pacientes tratados con AINE. El uso de diclofenaco puede atenuar los efectos CV de varios agentes terapéuticos utilizados para tratar estas afecciones médicas (p. ej., diuréticos, inhibidores de la ECA o bloqueadores de los receptores de angiotensina [BRA]) (ver INTERACCIONES CON LA DROGAS ).
Evite el uso de VOLTAREN en pacientes con insuficiencia cardíaca grave a menos que se espere que los beneficios superen el riesgo de empeoramiento de la insuficiencia cardíaca. Si se usa VOLTAREN 50 mg en pacientes con insuficiencia cardíaca grave, controle a los pacientes para detectar signos de empeoramiento de la insuficiencia cardíaca.
Toxicidad renal e hiperpotasemia
Toxicidad renal
La administración a largo plazo de NSAID ha resultado en necrosis papilar renal y otras lesiones renales.
También se ha observado toxicidad renal en pacientes en los que las prostaglandinas renales tienen un papel compensatorio en el mantenimiento de la perfusión renal. En estos pacientes, la administración de un AINE puede causar una reducción dependiente de la dosis en la formación de prostaglandinas y, secundariamente, en el flujo sanguíneo renal, lo que puede precipitar una descompensación renal manifiesta. Los pacientes con mayor riesgo de esta reacción son aquellos con función renal alterada, deshidratación, hipovolemia, insuficiencia cardíaca, disfunción hepática, aquellos que toman diuréticos e inhibidores de la ECA o BRA y los ancianos. La interrupción del tratamiento con AINE suele ir seguida de la recuperación al estado previo al tratamiento.
No hay información disponible de estudios clínicos controlados sobre el uso de VOLTAREN 100 mg en pacientes con enfermedad renal avanzada. Los efectos renales de VOLTAREN 50 mg pueden acelerar la progresión de la disfunción renal en pacientes con enfermedad renal preexistente.
Corrija el estado de volumen en pacientes deshidratados o hipovolémicos antes de iniciar VOLTAREN. Controle la función renal en pacientes con insuficiencia renal o hepática, insuficiencia cardíaca, deshidratación o hipovolemia durante el uso de VOLTAREN (ver INTERACCIONES CON LA DROGAS ). Evite el uso de VOLTAREN en pacientes con enfermedad renal avanzada a menos que se espere que los beneficios superen el riesgo de empeoramiento de la función renal. Si se usa VOLTAREN 50 mg en pacientes con enfermedad renal avanzada, controle a los pacientes para detectar signos de empeoramiento de la función renal.
hiperpotasemia
Se han informado aumentos en la concentración sérica de potasio, incluida la hiperpotasemia, con el uso de AINE, incluso en algunos pacientes sin insuficiencia renal. En pacientes con función renal normal, estos efectos se han atribuido a un estado de hiporreninemia-hipoaldosteronismo.
Reacciones anafilácticas
El diclofenaco se ha asociado con reacciones anafilácticas en pacientes con y sin hipersensibilidad conocida al diclofenaco y en pacientes con asma sensible a la aspirina (ver CONTRAINDICACIONES , ADVERTENCIAS ; Exacerbación del asma relacionada con la sensibilidad a la aspirina ).
Exacerbación del asma relacionada con la sensibilidad a la aspirina
Una subpoblación de pacientes con asma puede tener asma sensible a la aspirina, que puede incluir rinosinusitis crónica complicada con pólipos nasales; broncoespasmo severo, potencialmente fatal; y/o intolerancia a la aspirina y otros AINE. Debido a que se ha informado reactividad cruzada entre la aspirina y otros AINE en pacientes sensibles a la aspirina, VOLTAREN 100 mg está contraindicado en pacientes con esta forma de sensibilidad a la aspirina (ver CONTRAINDICACIONES ). Cuando se use VOLTAREN 100 mg en pacientes con asma preexistente (sin sensibilidad conocida a la aspirina), controle a los pacientes para detectar cambios en los signos y síntomas del asma.
Reacciones cutáneas graves
Los AINE, incluido el diclofenaco, pueden causar reacciones adversas graves en la piel, como dermatitis exfoliativa, síndrome de Stevens-Johnson (SJS) y necrólisis epidérmica tóxica (NET), que pueden ser fatales. Estos eventos graves pueden ocurrir sin previo aviso. Informar a los pacientes sobre los signos y síntomas de reacciones cutáneas graves y suspender el uso de VOLTAREN ante la primera aparición de erupción cutánea o cualquier otro signo de hipersensibilidad. VOLTAREN está contraindicado en pacientes con reacciones cutáneas graves previas a los AINE (ver CONTRAINDICACIONES ).
Reacción a medicamentos con eosinofilia y síntomas sistémicos (DRESS)
Se ha notificado una reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS) en pacientes que toman AINE, como VOLTAREN. Algunos de estos eventos han sido fatales o potencialmente mortales. DRESS normalmente, aunque no exclusivamente, se presenta con fiebre, erupción cutánea, linfadenopatía y/o hinchazón facial. Otras manifestaciones clínicas pueden incluir hepatitis, nefritis, anomalías hematológicas, miocarditis o miositis. A veces, los síntomas de DRESS pueden parecerse a una infección viral aguda. La eosinofilia a menudo está presente. Debido a que este trastorno es variable en su presentación, pueden estar involucrados otros sistemas de órganos que no se mencionan aquí. Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad, como fiebre o linfadenopatía, pueden estar presentes aunque la erupción no sea evidente. Si tales signos o síntomas están presentes, suspenda VOLTAREN 50 mg y evalúe al paciente de inmediato.
Toxicidad Fetal
Cierre prematuro del conducto arterioso fetal
Evite el uso de AINE, incluido VOLTAREN 50 mg, en mujeres embarazadas de aproximadamente 30 semanas de gestación y más. Los AINE, incluido VOLTAREN 50 mg, aumentan el riesgo de cierre prematuro del conducto arterioso fetal aproximadamente a esta edad gestacional.
Oligohidramnios/insuficiencia renal neonatal
El uso de AINE, incluido VOLTAREN, aproximadamente a las 20 semanas de gestación o más tarde en el embarazo puede causar disfunción renal fetal que conduce a oligohidramnios y, en algunos casos, insuficiencia renal neonatal. Estos resultados adversos se observan, en promedio, después de días o semanas de tratamiento, aunque el oligohidramnios se ha informado con poca frecuencia tan pronto como 48 horas después del inicio del AINE. El oligohidramnios a menudo, pero no siempre, es reversible con la interrupción del tratamiento. Las complicaciones del oligohidramnios prolongado pueden incluir, por ejemplo, contracturas de las extremidades y maduración pulmonar retrasada. En algunos casos posteriores a la comercialización de insuficiencia renal neonatal, se requirieron procedimientos invasivos, como exanguinotransfusión o diálisis.
Si el tratamiento con AINE es necesario entre las 20 y las 30 semanas de gestación, limite el uso de VOLTAREN a la dosis efectiva más baja y la duración más breve posible. Considere el control por ultrasonido del líquido amniótico si el tratamiento con VOLTAREN 50 mg se prolonga más de 48 horas. Suspender VOLTAREN 100 mg si se presenta oligohidramnios y realizar un seguimiento de acuerdo con la práctica clínica (ver PRECAUCIONES ; El embarazo ).
Toxicidad hematológica
Se ha producido anemia en pacientes tratados con AINE. Esto puede deberse a una pérdida de sangre oculta o macroscópica, retención de líquidos o un efecto descrito de forma incompleta sobre la eritropoyesis. Si un paciente tratado con VOLTAREN presenta algún signo o síntoma de anemia, controle la hemoglobina o el hematocrito.
Los AINE, incluido VOLTAREN 100 mg, pueden aumentar el riesgo de eventos hemorrágicos. Condiciones comórbidas, como trastornos de la coagulación, el uso concomitante de warfarina, otros anticoagulantes, agentes antiplaquetarios (p. ej., aspirina), inhibidores de la recaptación de serotonina (ISRS) e inhibidores de la recaptación de serotonina y norepinefrina (IRSN) pueden aumentar este riesgo. Vigile a estos pacientes en busca de signos de sangrado (ver INTERACCIONES CON LA DROGAS ).
PRECAUCIONES
General
No se puede esperar que VOLTAREN® (tabletas con cubierta entérica de diclofenaco sódico) sustituya a los corticosteroides o que trate la insuficiencia de corticosteroides. Una abrupta interrupción de los corticosteroides puede llevar a una exacerbación de la enfermedad. Los pacientes que reciben una terapia prolongada con corticosteroides deben reducir gradualmente su terapia si se toma la decisión de suspender los corticosteroides y se debe observar de cerca al paciente para detectar cualquier evidencia de efectos adversos, incluida la insuficiencia suprarrenal y la exacerbación de los síntomas de la artritis.
La actividad farmacológica de VOLTAREN 50 mg para reducir la fiebre y la inflamación puede disminuir la utilidad de estos signos diagnósticos para detectar complicaciones de presuntas afecciones dolorosas no infecciosas.

Información para pacientes
Aconseje al paciente que lea la etiqueta del paciente aprobada por la FDA ( Guía de medicamentos ) que acompaña a cada receta dispensada. Informe a los pacientes, familiares o cuidadores de la siguiente información antes de iniciar la terapia con VOLTAREN y periódicamente durante el transcurso de la terapia en curso.
Eventos trombóticos cardiovasculares
Aconseje a los pacientes que estén atentos a los síntomas de eventos trombóticos cardiovasculares, incluidos dolor torácico, dificultad para respirar, debilidad o dificultad para hablar, y que informen de inmediato cualquiera de estos síntomas a su proveedor de atención médica (ver ADVERTENCIAS ; Eventos trombóticos cardiovasculares ).
Sangrado gastrointestinal, ulceración y perforación
Aconseje a los pacientes que informen a su proveedor de atención médica sobre los síntomas de ulceraciones y sangrado, incluidos dolor epigástrico, dispepsia, melena y hematemesis. En el contexto del uso concomitante de aspirina en dosis bajas para la profilaxis cardíaca, informar a los pacientes sobre el aumento del riesgo de signos y síntomas de hemorragia GI (ver ADVERTENCIA ; Hemorragia gastrointestinal , Ulceración , y Perforación ).
hepatotoxicidad
Informar a los pacientes sobre los signos y síntomas de advertencia de hepatotoxicidad (p. ej., náuseas, fatiga, letargo, prurito, diarrea, ictericia, sensibilidad en el cuadrante superior derecho y síntomas “similares a la gripe”). Si esto ocurre, indique a los pacientes que suspendan VOLTAREN 50 mg y busquen tratamiento médico inmediato (ver ADVERTENCIAS ; hepatotoxicidad ).
Insuficiencia cardíaca y edema
Aconseje a los pacientes que estén atentos a los síntomas de insuficiencia cardíaca congestiva, que incluyen dificultad para respirar, aumento de peso inexplicable o edema, y que se comuniquen con su proveedor de atención médica si se presentan tales síntomas (ver ADVERTENCIAS ; Insuficiencia cardíaca y edema ).
Reacciones anafilácticas
Informe a los pacientes sobre los signos de una reacción anafiláctica (p. ej., dificultad para respirar, hinchazón de la cara o la garganta). Indique a los pacientes que busquen ayuda de emergencia inmediata si esto ocurre (ver ADVERTENCIAS ; Reacciones anafilácticas ).
Reacciones cutáneas graves, incluido DRESS
Aconseje a los pacientes que dejen de tomar VOLTAREN 50 mg de inmediato si desarrollan cualquier tipo de erupción o fiebre y que se comuniquen con su proveedor de atención médica lo antes posible (ver ADVERTENCIAS ; Reacciones cutáneas graves ).
Fertilidad Femenina
Aconseje a las mujeres con potencial reproductivo que deseen quedar embarazadas que los AINE, incluido VOLTAREN 50 mg, pueden estar asociados con un retraso reversible en la ovulación (ver PRECAUCIONES ; Carcinogénesis, mutagénesis, deterioro de la fertilidad ).
Toxicidad Fetal
Informar a las mujeres embarazadas que eviten el uso de VOLTAREN y otros AINE a partir de las 30 semanas de gestación por el riesgo de cierre prematuro del conducto arterioso fetal. Si se necesita tratamiento con VOLTAREN para una mujer embarazada entre las 20 y las 30 semanas de gestación, infórmele que es posible que deba ser monitoreada por oligohidramnios, si el tratamiento continúa por más de 48 horas. (ver ADVERTENCIAS ; Toxicidad Fetal , PRECAUCIONES , El embarazo ).
Evite el uso concomitante de AINE
Informe a los pacientes que no se recomienda el uso concomitante de VOLTAREN 50 mg con otros AINE o salicilatos (p. ej., diflunisal, salsalato) debido al mayor riesgo de toxicidad gastrointestinal y poco o ningún aumento de la eficacia (ver ADVERTENCIAS ; Hemorragia gastrointestinal , Ulceración , y Perforación e interacciones farmacológicas ). Alerte a los pacientes que los AINE pueden estar presentes en medicamentos de venta libre para el tratamiento de resfriados, fiebre o insomnio.
Uso de AINE y aspirina en dosis bajas
Informe a los pacientes que no usen aspirina en dosis bajas de forma concomitante con VOLTAREN hasta que hablen con su proveedor de atención médica (ver INTERACCIONES CON LA DROGAS ).
Enmascaramiento de inflamación y fiebre
La actividad farmacológica de VOLTAREN para reducir la inflamación y posiblemente la fiebre, puede disminuir la utilidad de los signos diagnósticos para detectar infecciones.
Monitoreo de laboratorio
Debido a que pueden ocurrir hemorragia gastrointestinal grave, hepatotoxicidad y daño renal sin síntomas o signos de advertencia, considere monitorear a los pacientes en tratamiento prolongado con AINE con un hemograma completo y un perfil químico periódicamente (ver ADVERTENCIAS ; Hemorragia gastrointestinal , Ulceración y Perforación , y hepatotoxicidad ).
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
Carcinogénesis
Los estudios de carcinogenicidad a largo plazo en ratas que recibieron diclofenaco sódico hasta 2 mg/kg/día (aproximadamente 0,1 veces la dosis humana máxima recomendada (MRHD) de VOLTAREN 50 mg, 200 mg/día, según la comparación del área de superficie corporal (BSA)) han revelado sin aumentos significativos en la incidencia de tumores. Un estudio de carcinogenicidad de 2 años realizado en ratones que emplearon diclofenaco sódico en dosis de hasta 0,3 mg/kg/día (aproximadamente 0,007 veces la MRHD según la comparación de BSA) en machos y 1 mg/kg/día (aproximadamente 0,02 veces la MRHD según la comparación de BSA) comparación de BSA) en mujeres no reveló ningún potencial oncogénico.
mutagénesis
El diclofenaco sódico no mostró actividad mutagénica en ensayos de mutación puntual in vitro en sistemas de prueba de mamíferos (linfoma de ratón) y microbianos (levadura, Ames) y no fue mutagénico en varias pruebas in vitro e in vivo de mamíferos, incluidos estudios de cromosomas epiteliales germinales masculinos y letales dominantes. en ratones, y estudios de anomalías del núcleo y aberraciones cromosómicas en hámsteres chinos.
Deterioro de la fertilidad
El diclofenaco sódico administrado a ratas macho y hembra en dosis de 4 mg/kg/día (aproximadamente 0,2 veces la MRHD según la comparación de BSA) no afectó la fertilidad.
Según el mecanismo de acción, el uso de AINE mediados por prostaglandinas, incluido VOLTAREN 100 mg, puede retrasar o prevenir la ruptura de los folículos ováricos, que se ha asociado con infertilidad reversible en algunas mujeres. Los estudios publicados en animales han demostrado que la administración de inhibidores de la síntesis de prostaglandinas tiene el potencial de interrumpir la ruptura folicular mediada por prostaglandinas necesaria para la ovulación. Pequeños estudios en mujeres tratadas con AINE también han mostrado un retraso reversible en la ovulación. Considere la suspensión de los AINE, incluido VOLTAREN 100 mg, en mujeres que tienen dificultades para concebir o que se encuentran bajo investigación por infertilidad.
El embarazo
Resumen de riesgos
El uso de AINE, incluido VOLTAREN, puede causar el cierre prematuro del conducto arterioso fetal y disfunción renal fetal que conduce a oligohidramnios y, en algunos casos, insuficiencia renal neonatal. Debido a estos riesgos, limite la dosis y la duración del uso de VOLTAREN entre las 20 y 30 semanas de gestación, y evite el uso de VOLTAREN alrededor de las 30 semanas de gestación y más adelante en el embarazo (ver ADVERTENCIAS ; Toxicidad Fetal ).
Cierre prematuro del conducto arterioso fetal
El uso de AINE, incluido VOLTAREN 100 mg, aproximadamente a las 30 semanas de gestación o más tarde en el embarazo aumenta el riesgo de cierre prematuro del conducto arterioso fetal.
Oligohidramnios/insuficiencia renal neonatal
El uso de AINE alrededor de las 20 semanas de gestación o más tarde en el embarazo se ha asociado con casos de disfunción renal fetal que conducen a oligohidramnios y, en algunos casos, insuficiencia renal neonatal.
No existen estudios adecuados y bien controlados de VOLTAREN 100 mg en mujeres embarazadas.
Los datos de los estudios observacionales sobre los posibles riesgos embriofetales del uso de AINE en mujeres en el primer o segundo trimestre del embarazo no son concluyentes.
En estudios de reproducción animal, no se observó evidencia de teratogenicidad en ratones, ratas o conejos que recibieron diclofenaco durante el período de organogénesis en dosis de hasta aproximadamente 0.5, 0.5 y 1 veces, respectivamente, la dosis máxima recomendada en humanos (MRHD) de VOLTAREN , 200 mg/día, a pesar de la presencia de toxicidad materna y fetal a estas dosis (ver Datos ).
Según los datos publicados en animales, se ha demostrado que las prostaglandinas tienen un papel importante en la permeabilidad vascular del endometrio, la implantación de blastocistos y la decidualización. En estudios con animales, la administración de inhibidores de la síntesis de prostaglandinas, como el diclofenaco, provocó un aumento de las pérdidas antes y después de la implantación. También se ha demostrado que las prostaglandinas tienen un papel importante en el desarrollo del riñón fetal. En estudios publicados en animales, se ha informado que los inhibidores de la síntesis de prostaglandinas afectan el desarrollo renal cuando se administran en dosis clínicamente relevantes.
Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo para la(s) población(es) indicada(s). Todos los embarazos tienen un riesgo de fondo de defecto congénito, pérdida u otros resultados adversos. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 % al 4 % y del 15 % al 20 %, respectivamente.
Consideraciones clínicas
Reacciones adversas fetales/neonatales
Cierre prematuro del conducto arterioso fetal:
Evite el uso de AINE en mujeres de aproximadamente 30 semanas de gestación y más adelante en el embarazo, porque los AINE, incluido VOLTAREN, pueden causar el cierre prematuro del conducto arterioso fetal (ver ADVERTENCIAS ; Toxicidad Fetal ).
Oligohidramnios/insuficiencia renal neonatal:
Si es necesario un AINE alrededor de las 20 semanas de gestación o más adelante en el embarazo, limite el uso a la dosis efectiva más baja y la duración más breve posible. Si el tratamiento con VOLTAREN 50 mg se extiende más allá de las 48 horas, considere monitorear con ultrasonido para detectar oligohidramnios. Si ocurre oligohidramnios, suspenda VOLTAREN y realice un seguimiento de acuerdo con la práctica clínica (ver ADVERTENCIAS ; Toxicidad Fetal ).
Datos
Datos humanos
Cierre prematuro del conducto arterioso fetal:
La literatura publicada informa que el uso de AINE alrededor de las 30 semanas de gestación y más adelante en el embarazo puede causar el cierre prematuro del conducto arterioso fetal.
Oligohidramnios/insuficiencia renal neonatal:
Los estudios publicados y los informes posteriores a la comercialización describen el uso materno de AINE alrededor de las 20 semanas de gestación o más tarde en el embarazo asociado con disfunción renal fetal que conduce a oligohidramnios y, en algunos casos, insuficiencia renal neonatal. Estos resultados adversos se observan, en promedio, después de días o semanas de tratamiento, aunque el oligohidramnios se ha informado con poca frecuencia tan pronto como 48 horas después del inicio del AINE. En muchos casos, pero no en todos, la disminución del líquido amniótico fue transitoria y reversible con el cese del fármaco. Ha habido un número limitado de informes de casos de uso materno de AINE y disfunción renal neonatal sin oligohidramnios, algunos de los cuales fueron irreversibles. Algunos casos de disfunción renal neonatal requirieron tratamiento con procedimientos invasivos, como exanguinotransfusión o diálisis.
Las limitaciones metodológicas de estos estudios e informes posteriores a la comercialización incluyen la falta de un grupo de control; información limitada sobre la dosis, la duración y el momento de la exposición al fármaco; y el uso concomitante de otros medicamentos. Estas limitaciones impiden establecer una estimación fiable del riesgo de resultados fetales y neonatales adversos con el uso materno de AINE. Debido a que los datos de seguridad publicados sobre los resultados neonatales involucraron principalmente a bebés prematuros, la generalización de ciertos riesgos informados para el bebé a término expuesto a AINE a través del uso materno es incierta.
Datos de animales
Los estudios de reproducción y desarrollo en animales demostraron que la administración de diclofenaco sódico durante la organogénesis no produjo teratogenicidad a pesar de la inducción de toxicidad materna y toxicidad fetal en ratones a dosis orales de hasta 20 mg/kg/día (aproximadamente 0,5 veces la dosis máxima recomendada en humanos [MRHD ] de VOLTAREN, 200 mg/día, con base en la comparación del área de superficie corporal (BSA), y en ratas y conejos en dosis orales de hasta 10 mg/kg/día (aproximadamente 0.5 y 1 veces, respectivamente, la MRHD con base en BSA comparación). En un estudio en el que a ratas preñadas se les administró por vía oral 2 o 4 mg/kg de diclofenaco (0,1 y 0,2 veces la MRHD según el BSA) desde el día 15 de gestación hasta el día 21 de lactancia, se observó toxicidad materna significativa (peritonitis, mortalidad). Estas dosis tóxicas para la madre se asociaron con distocia, gestación prolongada, peso y crecimiento fetal reducidos y supervivencia fetal reducida. Se ha demostrado que el diclofenaco atraviesa la barrera placentaria en ratones, ratas y humanos.
trabajo de parto o parto
No existen estudios sobre los efectos de VOLTAREN durante el trabajo de parto o el parto. En estudios con animales, los AINE, incluido el diclofenaco, inhiben la síntesis de prostaglandinas, retrasan el parto y aumentan la incidencia de muerte fetal.
Madres lactantes
Resumen de riesgos
Según los datos disponibles, el diclofenaco puede estar presente en la leche humana. Los beneficios para el desarrollo y la salud de la lactancia materna se deben considerar junto con la necesidad clínica de VOLTAREN de la madre y cualquier posible efecto adverso en el lactante a causa de VOLTAREN 100 mg o de la afección materna subyacente.
Datos
Una mujer tratada por vía oral con una sal de diclofenaco, 150 mg/día, tenía un nivel de diclofenaco en la leche de 100 mcg/L, equivalente a una dosis infantil de alrededor de 0,03 mg/kg/día. No se detectó diclofenaco en la leche materna en 12 mujeres que usaban diclofenaco (después de 100 mg/día por vía oral durante 7 días o de una dosis única intramuscular de 50 mg administrada en el período posparto inmediato).
Uso pediátrico
No se ha establecido la seguridad y eficacia en pacientes pediátricos.
Uso geriátrico
Los pacientes de edad avanzada, en comparación con los pacientes más jóvenes, tienen un mayor riesgo de reacciones adversas cardiovasculares, gastrointestinales y/o renales graves asociadas a los AINE. Si el beneficio anticipado para el paciente anciano supera estos riesgos potenciales, comience la dosificación en el extremo inferior del rango de dosificación y controle a los pacientes para detectar efectos adversos (ver ADVERTENCIAS ; Eventos trombóticos cardiovasculares , Gastrointestinal Hemorragia, Ulceración y Perforación, Hepatotoxicidad, Toxicidad Renal e Hiperpotasemia, PRECAUCIONES; Monitoreo de laboratorio).
Se sabe que el diclofenaco se excreta sustancialmente por vía renal, y el riesgo de reacciones adversas a este fármaco puede ser mayor en pacientes con insuficiencia renal. Debido a que es más probable que los pacientes de edad avanzada tengan una función renal disminuida, se debe tener cuidado al seleccionar la dosis y puede ser útil monitorear la función renal (ver FARMACOLOGÍA CLÍNICA , REACCIONES ADVERSAS ).
SOBREDOSIS
No se proporcionó información
CONTRAINDICACIONES
VOLTAREN® está contraindicado en los siguientes pacientes:
- Hipersensibilidad conocida (p. ej., reacciones anafilácticas y reacciones cutáneas graves) al diclofenaco o a cualquiera de los componentes del medicamento (ver ADVERTENCIAS ; Reacciones anafilácticas , Reacciones cutáneas graves ).
- Antecedentes de asma, urticaria u otras reacciones de tipo alérgico después de tomar aspirina u otros AINE. En estos pacientes se han notificado reacciones anafilácticas graves, a veces mortales, a los AINE (ver ADVERTENCIAS ; Reaccion anafiláctica , Exacerbación del asma relacionada con la sensibilidad a la aspirina ).
- En el marco de la cirugía de injerto de derivación de arteria coronaria (CABG) (ver ADVERTENCIAS ; Eventos trombóticos cardiovasculares ).
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
El diclofenaco tiene propiedades analgésicas, antiinflamatorias y antipiréticas.
El mecanismo de acción de VOLTAREN 100 mg, al igual que el de otros AINE, no se comprende por completo, pero implica la inhibición de la ciclooxigenasa (COX-1 y COX-2).
El diclofenaco es un potente inhibidor de la síntesis de prostaglandinas in vitro. Las concentraciones de diclofenaco alcanzadas durante la terapia han producido efectos in vivo. Las prostaglandinas sensibilizan los nervios aferentes y potencian la acción de la bradicinina en la inducción del dolor en modelos animales. Las prostaglandinas son mediadores de la inflamación. Dado que el diclofenaco es un inhibidor de la síntesis de prostaglandinas, su modo de acción puede deberse a una disminución de las prostaglandinas en los tejidos periféricos.
Farmacocinética
Absorción
El diclofenaco se absorbe al 100 % después de la administración oral en comparación con la administración intravenosa (IV), según lo medido por la recuperación de orina. Sin embargo, debido al metabolismo de primer paso, solo alrededor del 50% de la dosis absorbida está disponible sistémicamente (ver Tabla 1). Los alimentos no tienen un efecto significativo sobre el grado de absorción de diclofenaco. Sin embargo, suele haber un retraso en el inicio de la absorción de 1 a 4,5 horas y una reducción de los niveles plasmáticos máximos de menos del 20%.
Distribución
El volumen aparente de distribución (V/F) del diclofenaco sódico es de 1,4 l/kg.
El diclofenaco se une en más del 99 % a las proteínas séricas humanas, principalmente a la albúmina. La unión a proteínas séricas es constante en el rango de concentración (0,15 a 105 mcg/mL) alcanzado con las dosis recomendadas.
El diclofenaco se difunde dentro y fuera del líquido sinovial. La difusión hacia la articulación ocurre cuando los niveles plasmáticos son más altos que los del líquido sinovial, después de lo cual el proceso se invierte y los niveles del líquido sinovial son más altos que los niveles plasmáticos. No se sabe si la difusión en la articulación juega un papel en la eficacia del diclofenaco.
Eliminación
Metabolismo
Se han identificado cinco metabolitos de diclofenaco en plasma y orina humanos. Los metabolitos incluyen 4'Âhidroxi-, 5'-hidroxi-, 3'-hidroxi-, 4',5-dihidroxi- y 3'-hidroxi-4'-metoxidiclofenaco. El principal metabolito del diclofenaco, el 4'-hidroxi-diclofenaco, tiene una actividad farmacológica muy débil. La formación de 4'Âhidroxi-diclofenaco está mediada principalmente por CYP2C9. Tanto el diclofenaco como sus metabolitos oxidativos experimentan glucuronidación o sulfatación seguida de excreción biliar. La glucuronidación de acilo mediada por UGT2B7 y la oxidación mediada por CYP2C8 también pueden desempeñar un papel en el metabolismo del diclofenaco. CYP3A4 es responsable de la formación de metabolitos menores, 5-hidroxi- y 3'-hidroxi-diclofenaco. En pacientes con disfunción renal, las concentraciones máximas de los metabolitos 4'-hidroxi- y 5-hidroxi-diclofenaco fueron de aproximadamente el 50 % y el 4 % del compuesto original después de una dosis oral única en comparación con el 27 % y el 1 % en sujetos sanos normales.
Excreción
El diclofenaco se elimina a través del metabolismo y la posterior excreción urinaria y biliar de los conjugados de glucurónido y sulfato de los metabolitos. Se excreta poco o nada de diclofenaco libre inalterado en la orina. Aproximadamente el 65% de la dosis se excreta en la orina y aproximadamente el 35% en la bilis como conjugados de diclofenaco inalterado más metabolitos. Debido a que la eliminación renal no es una vía de eliminación importante para el diclofenaco inalterado, no es necesario ajustar la dosis en pacientes con disfunción renal de leve a moderada. La vida media terminal del diclofenaco inalterado es de aproximadamente 2 horas.
Poblaciones Especiales
Pediátrico
No se ha investigado la farmacocinética de VOLTAREN en pacientes pediátricos.
La raza
No se han identificado diferencias farmacocinéticas debidas a la raza.
Deterioro hepático
El metabolismo hepático representa casi el 100 % de la eliminación de 100 mg de VOLTAREN, por lo que los pacientes con enfermedad hepática pueden requerir dosis reducidas de 50 mg de VOLTAREN en comparación con los pacientes con una función hepática normal.
Insuficiencia renal
Se ha investigado la farmacocinética del diclofenaco en sujetos con insuficiencia renal. No se han detectado diferencias en la farmacocinética de diclofenaco en estudios de pacientes con insuficiencia renal. En pacientes con insuficiencia renal (aclaramiento de inulina de 60 a 90, de 30 a 60 y menos de 30 ml/min; N = 6 en cada grupo), los valores del área bajo la curva (AUC) y la tasa de eliminación fueron comparables a los de sujetos sanos .
Estudios de interacciones farmacológicas
Voriconazol
Cuando se coadministró con voriconazol (inhibidor de las enzimas CYP2C9, 2C19 y 3A4), la Cmáx y el AUC de diclofenaco aumentaron un 114 % y un 78 %, respectivamente (ver INTERACCIONES CON LA DROGAS ).
Aspirina
Cuando se administraron AINE con aspirina, se redujo la unión a proteínas de los AINE, aunque no se alteró la depuración de los AINE libres. Se desconoce el significado clínico de esta interacción. Consulte la Tabla 2 para conocer las interacciones farmacológicas clínicamente significativas de los AINE con la aspirina (consulte INTERACCIONES CON LA DROGAS ).
INFORMACIÓN DEL PACIENTE
Guía de medicamentos
VOLTAREN® (Vowl-tar-uhn) (diclofenaco sódico) comprimidos recubiertos
¿Cuál es la información más importante que debo saber sobre los medicamentos denominados antiinflamatorios no esteroideos (AINE)?
Los AINE pueden causar efectos secundarios graves, que incluyen:
- Mayor riesgo de sufrir un ataque al corazón o un derrame cerebral que puede conducir a la muerte. Este riesgo puede ocurrir temprano en el tratamiento y puede aumentar:
- con dosis crecientes de AINE
- con un uso prolongado de AINE
No tome AINE justo antes o después de una cirugía cardíaca llamada “injerto de derivación de arteria coronaria (CABG)”. Evite tomar AINE después de un ataque cardíaco reciente, a menos que su proveedor de atención médica se lo indique. Puede tener un mayor riesgo de sufrir otro ataque al corazón si toma AINE después de un ataque al corazón reciente.
- Mayor riesgo de sangrado, úlceras y desgarros (perforación) del esófago (conducto que va de la boca al estómago), estómago e intestinos:
- en cualquier momento durante el uso
- sin síntomas de advertencia
- que puede causar la muerte
El riesgo de desarrollar una úlcera o sangrado aumenta con:
- antecedentes de úlceras estomacales o sangrado estomacal o intestinal con el uso de AINE
- tomando medicamentos llamados "corticosteroides", "anticoagulantes", "ISRS" o "IRSN"
- dosis crecientes de AINE
- uso prolongado de AINE
- de fumar
- bebiendo alcohol
- mayor edad
- pobre salud
- enfermedad hepática avanzada
- problemas de sangrado
Los AINE solo deben usarse:
- exactamente como se prescribe
- a la dosis más baja posible para su tratamiento
- por el menor tiempo necesario
¿Qué son los AINE?
Los AINE se usan para tratar el dolor y el enrojecimiento, la hinchazón y el calor (inflamación) causados por afecciones médicas, como diferentes tipos de artritis, cólicos menstruales y otros tipos de dolor a corto plazo.
¿Quién no debe tomar AINE?
No tome AINE:
- si tuvo un ataque de asma, urticaria u otra reacción alérgica con aspirina o cualquier otro AINE.
- justo antes o después de la cirugía de derivación cardíaca.
Antes de tomar AINE, infórmele a su proveedor de atención médica sobre todas sus afecciones médicas, incluso si usted:
- tiene problemas hepáticos o renales
- tiene presión arterial alta
- tener asma
- está embarazada o planea quedar embarazada. Tomar NSAID alrededor de las 20 semanas de embarazo o más tarde puede dañar a su bebé por nacer. Si necesita tomar AINE durante más de 2 días cuando tiene entre 20 y 30 semanas de embarazo, es posible que su proveedor de atención médica deba controlar la cantidad de líquido en su útero alrededor de su bebé. No debe tomar AINE después de aproximadamente 30 semanas de embarazo.
- están amamantando o planean amamantar.
Informe a su proveedor de atención médica sobre todos los medicamentos que toma, incluidos los medicamentos recetados o de venta libre, las vitaminas o los suplementos a base de hierbas. Los AINE y algunos otros medicamentos pueden interactuar entre sí y causar efectos secundarios graves. No empiece a tomar ningún medicamento nuevo sin hablar primero con su proveedor de atención médica.
¿Cuáles son los posibles efectos secundarios de los AINE?
Los AINE pueden causar efectos secundarios graves, que incluyen:
Consulte "¿Cuál es la información más importante que debo saber sobre los medicamentos denominados antiinflamatorios no esteroideos (AINE)?"
- presión arterial alta nueva o peor
- insuficiencia cardiaca
- problemas hepáticos, incluida la insuficiencia hepática
- problemas renales, incluida la insuficiencia renal
- glóbulos rojos bajos (anemia)
- reacciones cutáneas potencialmente mortales
- reacciones alérgicas potencialmente mortales
- Otros efectos secundarios de los AINE incluyen: dolor de estómago, estreñimiento, diarrea, gases, acidez estomacal, náuseas, vómitos, mareos
Obtenga ayuda de emergencia de inmediato si tiene alguno de los siguientes síntomas:
- dificultad para respirar o dificultad para respirar
- Dolor de pecho
- debilidad en una parte o lado de su cuerpo
- dificultad para hablar
- hinchazón de la cara o la garganta
Deje de tomar su AINE y llame a su proveedor de atención médica de inmediato si presenta alguno de los siguientes síntomas:
- náuseas
- vomitar sangre
- más cansado o más débil de lo habitual
- Diarrea
- Comezón
- hay sangre en su evacuación intestinal o es negra y pegajosa como alquitrán
- tu piel o tus ojos se ven amarillos
- indigestión o dolor de estómago
- síntomas similares a la gripe
- aumento de peso inusual
- erupción cutánea o ampollas con fiebre
- hinchazón de brazos y piernas, manos y pies
Si toma demasiado AINE, llame a su proveedor de atención médica o busque ayuda médica de inmediato.
Estos no son todos los posibles efectos secundarios de los AINE. Para obtener más información, consulte a su proveedor de atención médica o farmacéutico acerca de los AINE. Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA1088
Otra información sobre los AINE.
- La aspirina es un medicamento AINE, pero no aumenta la posibilidad de sufrir un ataque al corazón. La aspirina puede causar sangrado en el cerebro, el estómago y los intestinos. La aspirina también puede causar úlceras en el estómago y los intestinos.
- Algunos AINE se venden en dosis más bajas sin receta (de venta libre). Hable con su proveedor de atención médica antes de usar AINE de venta libre durante más de 10 días.
Información general sobre el uso seguro y eficaz de los AINE.
veces, los medicamentos se recetan para fines distintos a los enumerados en la Guía del medicamento. No use NSAID para una condición para la cual no fue recetado. No le dé AINE a otras personas, incluso si tienen los mismos síntomas que usted. Puede que les haga daño.
Si desea obtener más información sobre los AINE, hable con su proveedor de atención médica. Puede pedirle a su farmacéutico o proveedor de atención médica información sobre los AINE escrita para profesionales de la salud.
Esta Guía del medicamento ha sido aprobada por la Administración de Alimentos y Medicamentos de EE. UU. Revisado: abril de 2021