Avapro 150mg, 300mg Irbesartan Uso, efectos secundarios, resistencia y dosis. Precio en farmacia online. Medicamentos genericos sin receta.
¿Qué es Avapro 300mg y cómo se usa?
Avapro 300 mg es un medicamento recetado que se usa para tratar los síntomas de la presión arterial alta (hipertensión), el dolor nervioso causado por la diabetes tipo 2. Avapro se puede usar solo o con otros medicamentos.
Avapro 300mg pertenece a una clase de medicamentos BRA.
No se sabe si Avapro 150 mg es seguro y efectivo en niños menores de 6 años.
¿Cuáles son los posibles efectos secundarios de Avapro 300mg?
Avapro puede causar efectos secundarios graves que incluyen:
- dolor muscular inexplicable,
- ternura o debilidad,
- fiebre,
- cansancio inusual,
- orina de color oscuro,
- aturdimiento,
- orinar poco o nada,
- hinchazón,
- rápido aumento de peso,
- confusión,
- pérdida de apetito,
- vómitos y
- dolor en el costado o en la parte baja de la espalda
Obtenga ayuda médica de inmediato si tiene alguno de los síntomas mencionados anteriormente.
Los efectos secundarios más comunes de Avapro incluyen:
- Diarrea,
- acidez,
- malestar estomacal y
- sensación de cansancio
Informe al médico si tiene algún efecto secundario que le moleste o que no desaparezca.
Estos no son todos los posibles efectos secundarios de Avapro. Para obtener más información, consulte a su médico o farmacéutico.
Llame a su médico para obtener asesoramiento médico sobre los efectos secundarios. Puede informar los efectos secundarios a la FDA al 1-800-FDA-1088.
ADVERTENCIA
TOXICIDAD FETAL
- Cuando se detecte un embarazo, suspenda AVAPRO lo antes posible.
- Los medicamentos que actúan directamente sobre el sistema renina-angiotens pueden causar lesiones y la muerte del feto en desarrollo [ver ADVERTENCIAS Y PRECAUCIONES].
DESCRIPCIÓN
AVAPRO (irbesartán) es un antagonista del receptor de la angiotensina II (subtipo AT1).
El irbesartán es un compuesto no peptídico, descrito químicamente como 2-butil-3-[p-(o-1H-tetrazol-5-ilfenil)bencil]-1,3-diazaspiro[4.4]non-1-en-4 -una.
Su fórmula empírica es C25H28N6O, y la fórmula estructural:
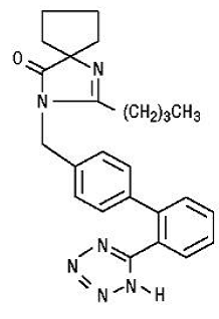
Irbesartán es un polvo cristalino de color blanco a blanquecino con un peso molecular de 428,5. Es un compuesto no polar con un coeficiente de reparto (octanol/agua) de 10,1 a un pH de 7,4. Irbesartán es ligeramente soluble en alcohol y cloruro de metileno y prácticamente insoluble en agua.
AVAPRO está disponible para administración oral en comprimidos sin ranurar que contienen 75 mg, 150 mg o 300 mg de irbesartán. Los ingredientes inactivos incluyen: lactosa, celulosa microcristalina, almidón pregelatinizado, croscarmelosa sódica, poloxámero 188, dióxido de silicio y estearato de magnesio.
INDICACIONES
Hipertensión
AVAPRO® está indicado para el tratamiento de la hipertensión, para bajar la presión arterial. Reducir la presión arterial reduce el riesgo de eventos cardiovasculares (CV) fatales y no fatales, principalmente accidentes cerebrovasculares e infarto de miocardio. Estos beneficios se han observado en ensayos controlados de fármacos antihipertensivos de una amplia variedad de clases farmacológicas, incluido este fármaco.
El control de la presión arterial alta debe ser parte del manejo integral del riesgo cardiovascular, incluido, según corresponda, el control de los lípidos, el manejo de la diabetes, la terapia antitrombótica, el abandono del hábito de fumar, el ejercicio y la ingesta limitada de sodio. Muchos pacientes requerirán más de 1 fármaco para lograr los objetivos de presión arterial. Para obtener consejos específicos sobre objetivos y manejo, consulte las pautas publicadas, como las del Comité Nacional Conjunto para la Prevención, Detección, Evaluación y Tratamiento de la Presión Arterial Alta (JNC, por sus siglas en inglés) del Programa Nacional de Educación sobre la Presión Arterial Alta.
Numerosos fármacos antihipertensivos, de una variedad de clases farmacológicas y con diferentes mecanismos de acción, han demostrado en ensayos controlados aleatorios que reducen la morbilidad y mortalidad cardiovascular, y se puede concluir que es la reducción de la presión arterial, y no alguna otra propiedad farmacológica de las drogas, que es en gran parte responsable de esos beneficios. El beneficio de resultado cardiovascular más grande y consistente ha sido una reducción en el riesgo de accidente cerebrovascular, pero también se han observado regularmente reducciones en el infarto de miocardio y la mortalidad cardiovascular.
La presión sistólica o diastólica elevada provoca un aumento del riesgo cardiovascular, y el aumento del riesgo absoluto por mmHg es mayor con presiones arteriales más altas, de modo que incluso reducciones modestas de la hipertensión grave pueden proporcionar un beneficio sustancial. La reducción del riesgo relativo de la reducción de la presión arterial es similar en poblaciones con riesgo absoluto variable, por lo que el beneficio absoluto es mayor en pacientes que tienen un riesgo más alto independientemente de su hipertensión (por ejemplo, pacientes con diabetes o hiperlipidemia), y se esperaría que tales pacientes beneficiarse de un tratamiento más agresivo para lograr un objetivo de presión arterial más bajo.
Algunos medicamentos antihipertensivos tienen efectos menores sobre la presión arterial (como monoterapia) en pacientes de raza negra, y muchos medicamentos antihipertensivos tienen indicaciones y efectos adicionales aprobados (p. ej., angina, insuficiencia cardíaca o nefropatía diabética). Estas consideraciones pueden guiar la selección de la terapia.
AVAPRO 300 mg puede usarse solo o en combinación con otros agentes antihipertensivos.
Nefropatía en pacientes diabéticos tipo 2
AVAPRO está indicado para el tratamiento de la nefropatía diabética en pacientes con diabetes tipo 2 e hipertensión, creatinina sérica elevada y proteinuria (>300 mg/día). En esta población, AVAPRO reduce la tasa de progresión de la nefropatía medida por la aparición de duplicación de la creatinina sérica o enfermedad renal en etapa terminal (necesidad de diálisis o trasplante renal) [ver Estudios clínicos ].
DOSIFICACIÓN Y ADMINISTRACIÓN
Consideraciones Generales
AVAPRO puede administrarse con otros agentes antihipertensivos y con o sin alimentos.
Hipertensión
La dosis inicial recomendada de AVAPRO 300 mg es de 150 mg una vez al día. La dosis se puede aumentar a una dosis máxima de 300 mg una vez al día según sea necesario para controlar la presión arterial [ver Estudios clínicos ].
Nefropatía en pacientes diabéticos tipo 2
La dosis recomendada es de 300 mg una vez al día [ver Estudios clínicos ].
Ajuste de dosis en pacientes con depleción de volumen y sal
La dosis inicial recomendada es de 75 mg una vez al día en pacientes con depleción del volumen intravascular o sal (p. ej., pacientes tratados enérgicamente con diuréticos o en hemodiálisis) [ver ADVERTENCIAS Y PRECAUCIONES ].
CÓMO SUMINISTRADO
Formas de dosificación y concentraciones
AVAPRO 75 mg es un comprimido recubierto con película de color blanco a blanquecino, biconvexo, ovalado, grabado con un corazón en un lado y “2871” en el otro.
AVAPRO 150 mg es un comprimido recubierto con película de color blanco a blanquecino, biconvexo, ovalado, grabado con un corazón en un lado y “2872” en el otro.
AVAPRO 300 mg es un comprimido recubierto con película de color blanco a blanquecino, biconvexo, ovalado, grabado con un corazón en un lado y “2873” en el otro.
Almacenamiento y manipulación
AVAPRO (irbesartán) está disponible en comprimidos recubiertos con película de color blanco a blanquecino, biconvexos, ovalados, grabados con una forma de corazón en un lado y un código en el otro (ver la tabla a continuación). Los frascos de unidad de uso contienen 30 o 90 comprimidos recubiertos con película de la siguiente manera:
Almacenar a 25°C (77°F); excursiones permitidas a 15°C–30°C (59°F–86°F) [ver Temperatura ambiente controlada por USP ].
sanofi-aventis US LLC, Bridgewater, NJ 08807, UNA EMPRESA DE SANOFI. Revisado: sep 2021
EFECTOS SECUNDARIOS
Las siguientes reacciones adversas importantes se describen en otra parte de la etiqueta:
- Hipotensión en pacientes con depleción de volumen o de sal [ver ADVERTENCIAS Y PRECAUCIONES ]
- Deterioro de la función renal [ver ADVERTENCIAS Y PRECAUCIONES ]
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se llevan a cabo en condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de los ensayos clínicos de otro fármaco y es posible que no reflejen las tasas observadas en la práctica. Sin embargo, la información sobre reacciones adversas de los ensayos clínicos proporciona una base para identificar los eventos adversos que parecen estar relacionados con el uso de medicamentos y para aproximar las tasas.
Hipertensión
Se evaluó la seguridad de AVAPRO en más de 4300 pacientes con hipertensión y alrededor de 5000 sujetos en general. Esta experiencia incluye 1303 pacientes tratados durante más de 6 meses y 407 pacientes durante 1 año o más.
En ensayos clínicos controlados con placebo, se informaron las siguientes reacciones adversas en al menos el 1 % de los pacientes tratados con AVAPRO (n=1965) y con una incidencia más alta en comparación con el placebo (n=641), excluyendo aquellas demasiado generales para ser informativas y aquellas no están razonablemente asociados con el uso del fármaco porque estaban asociados con la afección que se está tratando o son muy comunes en la población tratada, incluyen: diarrea (3 % frente a 2 %), dispepsia/ardor de estómago (2 % frente a 1 %) y fatiga (4% frente a 3%).
El uso de irbesartán no se asoció con un aumento de la incidencia de tos seca, como suele ocurrir con el uso de inhibidores de la ECA. En estudios controlados con placebo, la incidencia de tos en pacientes tratados con irbesartán fue del 2,8 % frente al 2,7 % en pacientes que recibieron placebo.
Nefropatía en pacientes diabéticos tipo 2
hiperpotasemia
En el ensayo de nefropatía diabética con irbesartán (IDNT) (proteinuria ≥900 mg/día y creatinina sérica que oscila entre 1,0 y 3,0 mg/dl), el porcentaje de pacientes con potasio >6 mEq/l fue del 18,6 % en el grupo de AVAPRO 300 mg versus 6,0% en el grupo placebo. Las interrupciones debidas a hiperpotasemia en el grupo de AVAPRO de 150 mg fueron del 2,1 % frente al 0,4 % en el grupo de placebo.
En IDNT, las reacciones adversas fueron similares a las observadas en pacientes con hipertensión, con la excepción de una mayor incidencia de síntomas ortostáticos que ocurrieron con mayor frecuencia en el grupo de AVAPRO 150 mg versus placebo: mareos (10,2 % frente a 6,0 %), mareos ortostáticos (5,4 % % vs 2,7%) e hipotensión ortostática (5,4% vs 3,2%).
Experiencia posterior a la comercialización
Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de AVAPRO. Debido a que estas reacciones son reportadas voluntariamente por una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
Urticaria; angioedema (que implica hinchazón de la cara, labios, faringe y/o lengua); reacción anafiláctica incluyendo shock anafiláctico; aumento de las pruebas de función hepática; ictericia; hepatitis; hiperpotasemia; anemia; trombocitopenia; aumento de CPK; tinnitus; e hipoglucemia en pacientes diabéticos.
INTERACCIONES CON LA DROGAS
Agentes que aumentan el potasio sérico
La coadministración de AVAPRO con otros medicamentos que elevan los niveles séricos de potasio puede provocar hiperpotasemia, a veces grave. Controle el potasio sérico en tales pacientes.
Litio
Se han informado aumentos en las concentraciones séricas de litio y toxicidad por litio con el uso concomitante de irbesartán y litio. Controle los niveles de litio en pacientes que reciben irbesartán y litio.
Medicamentos antiinflamatorios no esteroideos (AINE), incluidos los inhibidores selectivos de la ciclooxigenasa-2 (inhibidores de la COX-2)
En pacientes de edad avanzada, con depleción de volumen (incluidos los que reciben tratamiento con diuréticos) o con función renal comprometida, la coadministración de AINE, incluidos los inhibidores selectivos de la COX-2, con antagonistas de los receptores de la angiotensina II (incluido el irbesartán) puede provocar el deterioro de la función renal. , incluida la posible insuficiencia renal aguda. Estos efectos suelen ser reversibles. Monitoree la función renal periódicamente en pacientes que reciben tratamiento con irbesartán y AINE.
El efecto antihipertensivo de los antagonistas de los receptores de la angiotensina II, incluido el irbesartán, puede atenuarse con los AINE, incluidos los inhibidores selectivos de la COX-2.
Bloqueo dual del sistema renina-angiotensina (RAS)
El bloqueo dual del RAS con bloqueadores de los receptores de angiotensina, inhibidores de la ECA o aliskiren se asocia con un mayor riesgo de hipotensión, hiperpotasemia y cambios en la función renal (incluida la insuficiencia renal aguda) en comparación con la monoterapia. La mayoría de los pacientes que reciben la combinación de dos inhibidores de RAS no obtienen ningún beneficio adicional en comparación con la monoterapia. En general, evite el uso combinado de inhibidores de RAS. Controle de cerca la presión arterial, la función renal y los electrolitos en pacientes que toman AVAPRO 150 mg y otros agentes que afectan el RAS.
No coadministre aliskiren con AVAPRO en pacientes con diabetes. Evite el uso de aliskiren con AVAPRO en pacientes con insuficiencia renal (TFG
ADVERTENCIAS
Incluido como parte de la PRECAUCIONES sección.
PRECAUCIONES
Toxicidad Fetal
AVAPRO 150 mg puede causar daño fetal cuando se administra a una mujer embarazada. El uso de fármacos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre del embarazo reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal. El oligohidramnios resultante puede asociarse con hipoplasia pulmonar fetal y deformaciones esqueléticas. Los posibles efectos adversos neonatales incluyen hipoplasia craneal, anuria, hipotensión, insuficiencia renal y muerte. Cuando se detecte un embarazo, suspenda AVAPRO lo antes posible [ver Uso en poblaciones específicas ].
Hipotensión en volumen o pacientes con depleción de sal
En pacientes con un sistema renina-angiotensina activado, como pacientes con depleción de volumen o de sal (p. ej., aquellos que están siendo tratados con altas dosis de diuréticos), puede ocurrir hipotensión sintomática después de iniciar el tratamiento con AVAPRO. Corrija el volumen o la depleción de sal antes de la administración de AVAPRO 300 mg o use una dosis inicial más baja [ver DOSIFICACIÓN Y ADMINISTRACIÓN ].
Insuficiencia renal
Los cambios en la función renal, incluida la insuficiencia renal aguda, pueden ser causados por fármacos que inhiben el sistema renina-angiotensina. Los pacientes cuya función renal puede depender en parte de la actividad del sistema renina-angiotensina (p. ej., pacientes con estenosis de la arteria renal, enfermedad renal crónica, insuficiencia cardíaca grave o hipovolemia) pueden correr un riesgo particular de desarrollar insuficiencia renal aguda o muerte. en AVAPRO. Vigilar la función renal periódicamente en estos pacientes. Considere suspender o interrumpir el tratamiento en pacientes que desarrollen una disminución clínicamente significativa de la función renal con AVAPRO [ver INTERACCIONES CON LA DROGAS ].
Toxicología no clínica
Carcinogénesis, Mutagénesis, Deterioro De La Fertilidad
No se observó evidencia de carcinogenicidad cuando se administró irbesartán en dosis de hasta 500/1000 mg/kg/día (machos/hembras, respectivamente) en ratas y 1000 mg/kg/día en ratones hasta por 2 años. Para ratas macho y hembra, 500 mg/kg/día proporcionaron una exposición sistémica promedio a irbesartan (AUC 0-24 horas, ligado más no ligado) alrededor de 3 y 11 veces, respectivamente, la exposición sistémica promedio en humanos que recibieron la dosis humana máxima recomendada. dosis (MRHD) de 300 mg de irbesartan/día, mientras que 1000 mg/kg/día (administrados solo a mujeres) proporcionaron una exposición sistémica promedio de aproximadamente 21 veces la informada para humanos en la MRHD. Para ratones machos y hembras, 1000 mg/kg/día proporcionaron una exposición a irbesartan de aproximadamente 3 y 5 veces, respectivamente, la exposición humana a 300 mg/día.
Irbesartán no fue mutagénico en una serie de pruebas in vitro (prueba microbiana de Ames, prueba de reparación de ADN en hepatocitos de rata, ensayo de mutación genética directa de células de mamífero V79). Irbesartán dio negativo en varias pruebas de inducción de aberraciones cromosómicas (in vitro—ensayo de linfocitos humanos; in vivo—estudio de micronúcleos de ratón).
Irbesartán no tuvo efectos adversos sobre la fertilidad o el apareamiento de ratas macho o hembra en dosis orales ≤650 mg/kg/día, proporcionando la dosis más alta una exposición sistémica a irbesartán (AUC 0-24 horas, unido más no unido) aproximadamente 5 veces mayor que encontrado en humanos que reciben la MRHD de 300 mg/día.
Uso en poblaciones específicas
El embarazo
Resumen de riesgos
AVAPRO puede causar daño fetal cuando se administra a una mujer embarazada. El uso de fármacos que actúan sobre el sistema renina-angiotensina durante el segundo y tercer trimestre del embarazo reduce la función renal fetal y aumenta la morbilidad y muerte fetal y neonatal [ver Consideraciones clínicas ]. La mayoría de los estudios epidemiológicos que examinan las anomalías fetales después de la exposición al uso de antihipertensivos en el primer trimestre no han distinguido los fármacos que afectan al sistema renina-angiotensina de otros agentes antihipertensivos. Cuando se detecte un embarazo, suspenda AVAPRO 150 mg lo antes posible.
Todos los embarazos tienen un riesgo de fondo de defecto congénito, pérdida u otros resultados adversos, independientemente de la exposición al fármaco. En la población general de EE. UU., el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2 % al 4 % y del 15 % al 20 %, respectivamente.
Consideraciones clínicas
Riesgo materno y/o embriofetal asociado a la enfermedad
La hipertensión en el embarazo aumenta el riesgo materno de preeclampsia, diabetes gestacional, parto prematuro y complicaciones del parto (p. ej., necesidad de cesárea y hemorragia posparto). La hipertensión aumenta el riesgo fetal de restricción del crecimiento intrauterino y muerte intrauterina. Las mujeres embarazadas con hipertensión deben ser monitoreadas cuidadosamente y tratadas en consecuencia.
Reacciones adversas fetales/neonatales
El oligohidramnios en mujeres embarazadas que usan medicamentos que afectan el sistema renina-angiotensina en el segundo y tercer trimestre del embarazo puede provocar lo siguiente: función renal fetal reducida que conduce a anuria e insuficiencia renal, hipoplasia pulmonar fetal, deformaciones esqueléticas, incluida hipoplasia del cráneo, hipotensión y muerte.
En el caso inusual de que no exista una alternativa adecuada a la terapia con medicamentos que afectan el sistema renina-angiotensina para un paciente en particular, informe a la madre del riesgo potencial para el feto.
Realice exámenes de ultrasonido en serie para evaluar el entorno intraamniótico. Las pruebas fetales pueden ser apropiadas, según la semana de embarazo. Sin embargo, los pacientes y los médicos deben ser conscientes de que es posible que el oligohidramnios no aparezca hasta después de que el feto haya sufrido una lesión irreversible. Si se observa oligohidramnios, considere un tratamiento alternativo. Observe de cerca a los bebés con antecedentes de exposición en el útero a AVAPRO para detectar hipotensión, oliguria e hiperpotasemia y otros síntomas de insuficiencia renal. En neonatos con antecedentes de exposición intrauterina a AVAPRO 300 mg, si se presenta oliguria o hipotensión, dirija la atención hacia el apoyo de la presión arterial y la perfusión renal. Puede requerirse transfusión de intercambio o diálisis como medio para revertir la hipotensión y/o sustituir la función renal alterada.
Datos
Datos de animales
Irbesartán atraviesa la placenta en ratas y conejos. En ratas hembras que recibieron irbesartán antes del apareamiento durante la gestación y la lactancia en dosis orales de 50, 180 o 650 mg/kg/día (1,6 a 21,1 veces la dosis máxima recomendada en humanos (MRHD) según el área de superficie corporal), los fetos examinados en El día 20 de gestación mostró una mayor incidencia de hidrouréter y cavitación pélvica renal y/o ausencia de papila renal en todos los grupos tratados con irbesartán. También se produjo edema subcutáneo en fetos con dosis maternas ≥180 mg/kg/día (5,8 veces la MRHD). Estas anomalías ocurrieron cuando las ratas hembra recibieron irbesartán desde antes del apareamiento hasta el día 20 de gestación, pero no se observaron en las crías después del nacimiento en el mismo estudio, o cuando se administró irbesartán a ratas preñadas solo durante la organogénesis (día de gestación 6 a día de gestación 15) en dosis orales de 50 a 450 mg/kg/día (hasta 14,6 veces la MRHD). Además, no se observaron efectos adversos sobre el desarrollo renal en cachorros de madres que recibieron irbesartán desde el día 15 de gestación hasta el día 24 de lactancia en dosis de 50, 180 o 650 mg/kg/día (hasta 21,1 veces la MRHD). Se cree que los efectos observados son efectos gestacionales tardíos de la droga. Las conejas preñadas que recibieron dosis orales de irbesartán de 30 mg/kg/día (1,9 veces la MRHD según el área de superficie corporal) experimentaron una alta tasa de mortalidad materna y aborto. Las hembras sobrevivientes tuvieron un ligero aumento en las reabsorciones tempranas y una disminución correspondiente en los fetos vivos.
La radioactividad estuvo presente en los fetos de ratas y conejos durante la última etapa de la gestación luego de dosis orales de irbesartán radiomarcado.
Lactancia
No hay datos disponibles sobre la presencia de irbesartán en la leche humana, los efectos sobre la producción de leche o el lactante. Irbesartán o algún metabolito de irbesartán se secreta en la leche de ratas lactantes [ver FARMACOLOGÍA CLÍNICA ]. Debido al potencial de efectos adversos en el lactante, no se recomienda el uso de AVAPRO 150 mg en mujeres lactantes.
Uso pediátrico
Irbesartan, en un estudio a una dosis de hasta 4,5 mg/kg/día, una vez al día, no pareció reducir la presión arterial de manera efectiva en pacientes pediátricos de 6 a 16 años.
AVAPRO no ha sido estudiado en pacientes pediátricos menores de 6 años.
Uso geriátrico
De 4925 sujetos que recibieron AVAPRO en estudios clínicos controlados de hipertensión, 911 (18,5 %) tenían 65 años o más, mientras que 150 (3,0 %) tenían 75 años o más. No se observaron diferencias generales en la eficacia o seguridad entre estos sujetos y los sujetos más jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores. [Ver FARMACOLOGÍA CLÍNICA y Estudios clínicos ]
SOBREDOSIS
No hay datos disponibles con respecto a la sobredosis en humanos. Sin embargo, las dosis diarias de 900 mg durante 8 semanas fueron bien toleradas. Se espera que las manifestaciones más probables de sobredosis sean hipotensión y taquicardia; la bradicardia también puede ocurrir por sobredosis. Irbesartán no se elimina por hemodiálisis.
Los estudios de toxicidad oral aguda con irbesartán en ratones y ratas indicaron que las dosis letales agudas superaron los 2000 mg/kg, aproximadamente 25 y 50 veces la MRHD (300 mg) según el área de superficie corporal, respectivamente.
CONTRAINDICACIONES
AVAPRO 150mg está contraindicado en pacientes hipersensibles a cualquiera de los componentes de este producto.
No coadministrar aliskireno con AVAPRO 300 mg en pacientes con diabetes.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción
La angiotensina II es un potente vasoconstrictor formado a partir de la angiotensina I en una reacción catalizada por la enzima convertidora de angiotensina (ACE, cininasa II). La angiotensina II es la principal hormona vasoactiva del sistema renina-angiotensina y un componente importante en la fisiopatología de la hipertensión. También estimula la secreción de aldosterona por la corteza suprarrenal. Irbesartán bloquea los efectos vasoconstrictores y secretores de aldosterona de la angiotensina II al unirse selectivamente al receptor AT1 de angiotensina II que se encuentra en muchos tejidos (p. ej., músculo liso vascular, glándula suprarrenal). También hay un receptor AT2 en muchos tejidos, pero no está involucrado en la homeostasis cardiovascular.
Irbesartán es un antagonista competitivo específico de los receptores AT1 con una afinidad mucho mayor (más de 8500 veces) por el receptor AT1 que por el receptor AT2 y sin actividad agonista.
El bloqueo del receptor AT1 elimina la retroalimentación negativa de la angiotensina II sobre la secreción de renina, pero el aumento resultante de la actividad de la renina plasmática y la angiotensina II circulante no superan los efectos del irbesartán sobre la presión arterial.
Irbesartan no inhibe la ECA o la renina ni afecta a otros receptores hormonales o canales iónicos que se sabe que están involucrados en la regulación cardiovascular de la presión arterial y la homeostasis del sodio.
Farmacodinámica
En sujetos sanos, dosis orales únicas de irbesartán de hasta 300 mg produjeron una inhibición dependiente de la dosis del efecto presor de las infusiones de angiotensina II. La inhibición fue completa (100 %) 4 horas después de dosis orales de 150 mg o 300 mg y la inhibición parcial se mantuvo durante 24 horas (60 % y 40 % con 300 mg y 150 mg, respectivamente).
En pacientes hipertensos, la inhibición del receptor de angiotensina II después de la administración crónica de irbesartán provoca un aumento de 1,5 a 2 veces en la concentración plasmática de angiotensina II y un aumento de 2 a 3 veces en los niveles de renina plasmática. Las concentraciones plasmáticas de aldosterona generalmente disminuyen después de la administración de irbesartán, pero los niveles séricos de potasio no se ven afectados significativamente a las dosis recomendadas.
En pacientes hipertensos, las dosis orales crónicas de irbesartán (hasta 300 mg) no tuvieron efecto sobre la tasa de filtración glomerular, el flujo plasmático renal o la fracción de filtración. En estudios de dosis múltiples en pacientes hipertensos, no hubo efectos clínicamente importantes sobre los triglicéridos en ayunas, el colesterol total o las concentraciones de colesterol HDL. No hubo efecto sobre el ácido úrico sérico durante la administración oral crónica, ni efecto uricosúrico.
Farmacocinética
Absorción
La absorción oral de irbesartan es rápida y completa con una biodisponibilidad absoluta promedio de 60% a 80%. Después de la administración oral de AVAPRO, las concentraciones plasmáticas máximas de irbesartán se alcanzan entre 1,5 y 2 horas después de la administración. Los alimentos no afectan la biodisponibilidad de irbesartán.
Irbesartán muestra una farmacocinética lineal en el rango de dosis terapéuticas.
Distribución
Irbesartan se une en un 90% a las proteínas séricas (principalmente albúmina y glicoproteína ácida α) con una unión insignificante a los componentes celulares de la sangre. El volumen medio de distribución es de 53 a 93 litros.
Los estudios en animales indican que el irbesartán radiomarcado atraviesa débilmente la barrera hematoencefálica y la placenta. Irbesartán se excreta en la leche de ratas lactantes.
Eliminación
Las depuraciones plasmáticas y renales totales están en el rango de 157 a 176 ml/min y de 3,0 a 3,5 ml/min, respectivamente. La vida media de eliminación terminal de irbesartán tiene un promedio de 11 a 15 horas. Las concentraciones en estado estacionario se alcanzan en 3 días. Se observa una acumulación limitada de irbesartan (
Metabolismo
Irbesartan es un agente oralmente activo que no requiere biotransformación en una forma activa. Irbesartán se metaboliza por conjugación y oxidación con glucurónidos. Después de la administración oral o intravenosa de Clabeled irbesartán, más del 80 % de la radiactividad plasmática circulante es atribuible al irbesartán inalterado. El principal metabolito circulante es el conjugado de glucurónido de irbesartan inactivo (aproximadamente 6%). Los metabolitos oxidativos restantes no se suman de manera apreciable a la actividad farmacológica de irbesartán.
Los estudios in vitro indican que irbesartan es oxidado principalmente por CYP2C9; el metabolismo por CYP3A4 es insignificante.
Excreción
Irbesartán y sus metabolitos se excretan tanto por vía biliar como renal. Después de la administración oral o intravenosa de irbesartan marcado con 14C, aproximadamente el 20% de la radioactividad se recupera en la orina y el resto en las heces, como irbesartán o glucurónido de irbesartán.
Poblaciones Específicas
Sexo
No se observaron diferencias relacionadas con el sexo en la farmacocinética en sujetos sanos de edad avanzada (edad 65-80 años) o en sujetos sanos jóvenes (edad 18-40 años). En estudios de pacientes hipertensos, no hay diferencia de sexo en la vida media o la acumulación, pero se observan concentraciones plasmáticas algo más altas de irbesartan en mujeres (11%-44%). No es necesario ajustar la dosis según el sexo.
Geriatría
En sujetos de edad avanzada (65 a 80 años de edad), la vida media de eliminación de irbesartán no se altera significativamente, pero los valores de AUC y Cmax son entre un 20 % y un 50 % mayores que los de los sujetos jóvenes (18 a 40 años de edad). No es necesario ajustar la dosis en los ancianos.
Raza/etnicidad
En sujetos sanos de raza negra, los valores de AUC de irbesartán son aproximadamente un 25 % mayores que en sujetos de raza blanca; no hay diferencia en los valores de Cmax.
Insuficiencia renal
La farmacocinética de irbesartán no se altera en pacientes con insuficiencia renal o en pacientes en hemodiálisis. Irbesartán no se elimina por hemodiálisis. No es necesario ajustar la dosis en pacientes con insuficiencia renal de leve a grave, a menos que un paciente con insuficiencia renal también tenga depleción de volumen [ver ADVERTENCIAS Y PRECAUCIONES y DOSIFICACIÓN Y ADMINISTRACIÓN ].
insuficiencia hepática
La farmacocinética de irbesartán después de la administración oral repetida no se ve afectada significativamente en pacientes con cirrosis hepática leve a moderada. No es necesario ajustar la dosis en pacientes con insuficiencia hepática.
Interacciones fármaco-fármaco
Los estudios in vitro muestran una inhibición significativa de la formación de metabolitos de irbesartán oxidados con los sustratos/inhibidores conocidos del citocromo CYP2C9 sulfenazol, tolbutamida y nifedipino. Sin embargo, en los estudios clínicos, las consecuencias de irbesartán concomitante sobre la farmacodinámica de la warfarina fueron insignificantes. Según los datos in vitro, no se esperaría interacción con medicamentos cuyo metabolismo depende de las isoenzimas 1A1, 1A2, 2A6, 2B6, 2D6, 2E1 o 3A4 del citocromo P450.
En estudios separados de pacientes que recibieron dosis de mantenimiento de warfarina, hidroclorotiazida o digoxina, la administración de irbesartán durante 7 días no tuvo efecto sobre la farmacodinámica de la warfarina (tiempo de protrombina) o la farmacocinética de la digoxina. La farmacocinética de irbesartán no se ve afectada por la coadministración de nifedipina o hidroclorotiazida.
Estudios clínicos
Hipertensión
Los efectos antihipertensivos de AVAPRO se examinaron en 7 ensayos controlados con placebo de 8 a 12 semanas en pacientes con presión arterial diastólica inicial de 95 a 110 mmHg. En estos ensayos se incluyeron dosis de 1 a 900 mg para explorar completamente el rango de dosis de irbesartán. Estos estudios permitieron la comparación de regímenes de una o dos veces al día a 150 mg/día, comparaciones de los efectos máximo y mínimo, y comparaciones de respuesta por sexo, edad y raza. Dos de los siete ensayos controlados con placebo identificados anteriormente examinaron los efectos antihipertensivos de irbesartán e hidroclorotiazida en combinación.
Los 7 estudios de monoterapia con irbesartan incluyeron un total de 1915 pacientes aleatorizados a irbesartan (1-900 mg) y 611 pacientes aleatorizados a placebo. Dosis una vez al día de 150 mg y 300 mg proporcionaron disminuciones estadística y clínicamente significativas en la presión arterial sistólica y diastólica con efectos mínimos (24 horas después de la dosis) después de 6 a 12 semanas de tratamiento en comparación con el placebo, de aproximadamente 8-10/ 5-6 mmHg y 8-12/5-8 mmHg, respectivamente. No se observó un mayor aumento del efecto con dosis superiores a 300 mg. Las relaciones dosis-respuesta para los efectos sobre la presión sistólica y diastólica se muestran en las Figuras 1 y 2.
Figura 1: Reducción sustraída con placebo en la SeSBP mínima; análisis integrado
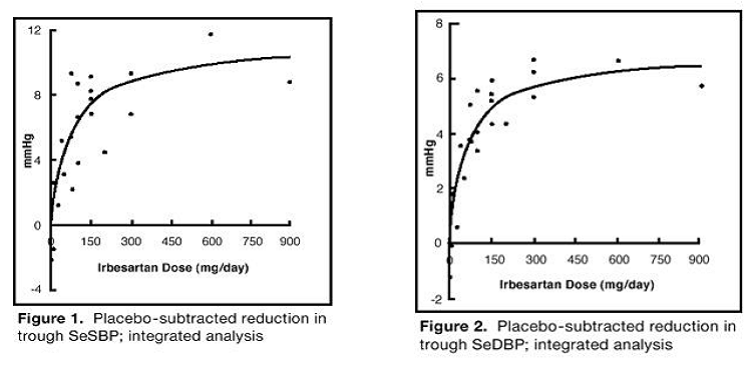
Figura 2: Reducción sustraída con placebo en la SeSBP mínima; análisis integrado
La administración una vez al día de dosis terapéuticas de irbesartán produjo efectos máximos alrededor de las 3 a 6 horas y, en un estudio de monitoreo ambulatorio de la presión arterial, nuevamente alrededor de las 14 horas. Esto se observó con la dosificación una y dos veces al día. Las proporciones de valle a pico para la respuesta sistólica y diastólica fueron generalmente entre 60% y 70%. En un estudio de monitoreo ambulatorio continuo de la presión arterial, la administración de 150 mg una vez al día proporcionó respuestas mínimas y medias de 24 horas similares a las observadas en pacientes que recibieron la misma dosis diaria total dos veces al día.
En ensayos controlados, la adición de irbesartán a dosis de hidroclorotiazida de 6,25 mg, 12,5 mg o 25 mg produjo reducciones adicionales relacionadas con la dosis en la presión arterial similares a las logradas con la misma dosis de monoterapia de irbesartán. HCTZ también tuvo un efecto aproximadamente aditivo.
El análisis de los subgrupos de edad, sexo y raza de los pacientes mostró que los hombres y las mujeres, y los pacientes mayores y menores de 65 años, tenían respuestas generalmente similares. Irbesartan fue eficaz para reducir la presión arterial independientemente de la raza, aunque el efecto fue algo menor en los negros (por lo general, una población con bajo nivel de renina).
El efecto de irbesartán es evidente después de la primera dosis, y está cerca de su efecto total observado a las 2 semanas. Al final de una exposición de 8 semanas, alrededor de 2/3 del efecto antihipertensivo seguía presente una semana después de la última dosis. No se observó hipertensión de rebote. Esencialmente, no hubo cambios en la frecuencia cardíaca promedio en pacientes tratados con irbesartán en ensayos controlados.
Nefropatía en pacientes diabéticos tipo 2
El Irbesartan Diabetic Nephropathy Trial (IDNT) fue un estudio multicéntrico, aleatorizado, controlado con placebo y activo, doble ciego, realizado en todo el mundo en 1715 pacientes con diabetes tipo 2, hipertensión (PAS Se > 135 mmHg o PAD Se > 85 mm Hg) y nefropatía creatinina de 1,0 a 3,0 mg/dl en mujeres o de 1,2 a 3,0 mg/dl en hombres y proteinuria ≥900 mg/día). Los pacientes fueron aleatorizados para recibir 75 mg de AVAPRO, 2,5 mg de amlodipino o un placebo equivalente una vez al día. Los pacientes recibieron una dosis de mantenimiento de 300 mg de AVAPRO o 10 mg de amlodipino, según la tolerancia. Se agregaron agentes antihipertensivos adicionales (excluidos los inhibidores de la ECA, los antagonistas de los receptores de la angiotensina II y los bloqueadores de los canales de calcio) según fue necesario para lograr el objetivo de presión arterial (≤135/85 o 10 mmHg de reducción de la presión arterial sistólica si es superior a 160 mmHg) para los pacientes de todos los grupos .
La población del estudio era 66,5 % masculina, 72,9 % menor de 65 años y 72 % blanca (5,0 % asiática/isleña del Pacífico, 13,3 % negra, 4,8 % hispana). Las presiones arteriales sistólica y diastólica basales medias en sedestación fueron de 159 mmHg y 87 mmHg, respectivamente. Los pacientes ingresaron al ensayo con una creatinina sérica media de 1,7 mg/dL y una proteinuria media de 4144 mg/día.
La presión arterial media alcanzada fue de 142/77 mmHg para AVAPRO, 142/76 mmHg para amlodipino y 145/79 mmHg para placebo. En general, el 83,0 % de los pacientes recibieron la dosis objetivo de irbesartán más del 50 % de las veces. Los pacientes fueron seguidos durante una duración media de 2,6 años.
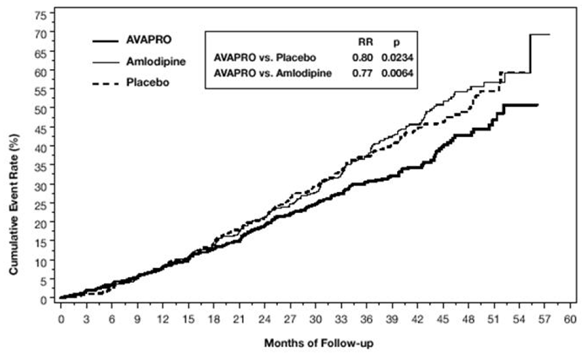
El criterio principal de valoración compuesto fue el tiempo hasta la aparición de cualquiera de los siguientes eventos: duplicación de la creatinina sérica inicial, enfermedad renal en etapa terminal (ESRD; definida por creatinina sérica ≥6 mg/dl, diálisis o trasplante renal) o muerte . El tratamiento con AVAPRO 150 mg resultó en una reducción del riesgo del 20 % en comparación con el placebo (p = 0,0234) (consulte la Figura 3 y la Tabla 1). El tratamiento con AVAPRO 150 mg también redujo la aparición de una duplicación sostenida de la creatinina sérica como un criterio de valoración separado (33 %), pero no tuvo un efecto significativo solo en la ESRD ni en la mortalidad general (consulte la Tabla 1).
Figura 3: IDNT: Estimaciones de Kaplan-Meier del criterio principal de valoración (duplicación de la creatinina sérica, enfermedad renal en etapa terminal o mortalidad por todas las causas)
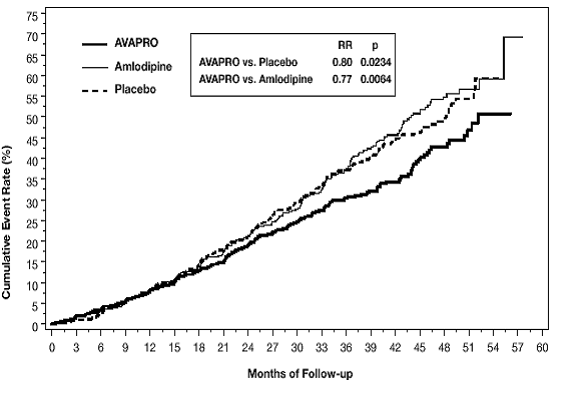
Los porcentajes de pacientes que experimentaron un evento durante el curso del estudio se pueden ver en la Tabla 1 a continuación:
El objetivo secundario del estudio fue un compuesto de mortalidad y morbilidad cardiovascular (infarto de miocardio, hospitalización por insuficiencia cardíaca, accidente cerebrovascular con déficit neurológico permanente, amputación). No hubo diferencias estadísticamente significativas entre los grupos de tratamiento en estos criterios de valoración. En comparación con el placebo, AVAPRO 150 mg redujo significativamente la proteinuria en aproximadamente un 27 %, un efecto que fue evidente dentro de los 3 meses posteriores al inicio de la terapia. AVAPRO redujo significativamente la tasa de pérdida de la función renal (tasa de filtración glomerular), medida por el recíproco de la concentración de creatinina sérica, en un 18,2 %.
La Tabla 2 presenta los resultados por subgrupos demográficos. Los análisis de subgrupos son difíciles de interpretar y no se sabe si estas observaciones representan diferencias verdaderas o efectos aleatorios. Para el criterio principal de valoración, los efectos favorables de AVAPRO se observaron en pacientes que también tomaban otros medicamentos antihipertensivos (no se permitieron los antagonistas de los receptores de angiotensina II, los inhibidores de la enzima convertidora de angiotensina y los bloqueadores de los canales de calcio), agentes hipoglucemiantes orales y antihipertensivos. agentes reductores.
INFORMACIÓN DEL PACIENTE
El embarazo
Asesorar a las pacientes en edad fértil sobre las consecuencias de la exposición a AVAPRO durante el embarazo. Analice las opciones de tratamiento con las mujeres que planean quedar embarazadas. Se debe pedir a los pacientes que informen de los embarazos a sus médicos lo antes posible.
Suplementos de potasio
Aconseje a los pacientes que reciben AVAPRO que no usen suplementos de potasio o sustitutos de la sal que contengan potasio sin consultar a su proveedor de atención médica [ver INTERACCIONES CON LA DROGAS ].